CMC Considerations when a Drug Development Project is Assigned Breakthrough Therapy Status

This article discusses the impact on Chemistry, Manufacturing and Control (CMC) part of a development project when a project is assigned Breakthrough Therapy (BT) status as given in Food and Drug Administration Safety and Innovation Act (FDASIA)1 and FDA Guidance on Expedited Programs for Serious Conditions.2
Assignment of Breakthrough Therapy (BT) designation could lead to accelerated clinical programs, which could be two or more years less than a “conventional” development program. Potential accelerated clinical development timelines could lead to insufficient time to complete all “traditional” CMC studies for approval and delivery to the patient within the boundaries of completing the clinical development program, for example:
- May have reduced real time stability for commercial material and need to leverage stability information from development studies
- Likely to have limited manufacturing experience at commercial scale, which presents the opportunity to leverage life cycle validation principles
- May need to consider launch with initial commercial supplies from a clinical manufacturing facility with clinical fit-for-purpose formulations and then convert over to a commercial formulation and plant immediately postapproval.
- The formulation and process could be ready for transfer, but the commercial facility is unavailable or not ready.
- Limited data sets from which to derive specification acceptance criteria
Using hypothetical case studies based on actual CMC development programs, a series of potential scenarios are given which could lead to discussions with the FDA. These case studies highlight from the overall development program the origins of the potential CMC challenges listed above. Discussions with the Agency should balance the risk of having less traditional CMC data at the time of filing with the potential benefit of a speedy delivery of critical product to patients. Regulatory approaches are proposed to address the lack of some “traditional” CMC data at the time of filing by:
- Employing more flexible filing processes; such as leveraging developmental data and risk assessments in lieu of some commercial scale experience.
- Using post-approval life-cycle management plans
- Including more comparability protocols in NDA submissions
- Employing more interaction opportunities with the Agency
Considerations are also given regarding facilitating interactions between a sponsor and FDA during a breakthrough therapy CMC development.
Introduction
On 9 July 2012, the Food and Drug Administration Safety and Innovation Act (FDASIA) was enacted in the US, which created the breakthrough therapy designation for promising new drugs that demonstrate substantial improvement over existing therapies for a serious or life-threatening disease in early clinical studies. The Breakthrough Therapy (BT) designation created an additional regulatory process for the FDA to expedite the development and commercial approval of drugs intended to treat a “serious disease or condition.” Other existing regulatory processes available to the FDA include accelerated approval, fast track and priority review. Table A is provided below to identify the primary criteria for each category and its impact on pharmaceutical development and regulatory review.2
The FDA’s breakthrough therapies pathway focuses on accelerating the existing approval process by having sponsors work closely with the Agency to develop trial designs that shorten or combine traditional phases of drug development. Other regulatory authorities are also considering programs for speeding access of promising new drugs to patients. In March 2014, the European Medicines Agency (EMA) announced an adaptive licensing pilot program3 which will use regulatory processes within the existing EU legal framework. In April 2014, the UK Agency, the Medicines and Healthcare products Regulatory Agency (MHRA) announced an early access to medicines scheme4 to support access to unlicensed medicines in areas of unmet medical need; and the Japanese regulatory agency, Ministry of Health, Labor and Welfare (MHLW) is considering introducing an accelerated regulatory processes to make promising new drugs available as quickly as possible to patients.
- 2FDA Guidance for Industry: Expedited Programs for Serious Conditions – Drugs and Biologics, June 2013, U.S. Food and Drug Administration (FDA), www.fda.gov.
- 3EMA/254350/2012, Pilot Project on Adaptive Licensing, 19 March 2014, European Medicines Agency (EMA), www.ema.europa.eu.
- 4Early Access to Medicines Scheme, http://www.mhra.gov.uk/NewsCentre/Pressreleases/CON404193, 7 April2014.
| Category | Applies to | Impact to Pharmaceutical Development and Regulatory Approval |
|---|---|---|
| Accelerated Approval Pathway |
Allowance to use a surrogate endpoint in clinical trials for initial approval in disease states with a substantial unmet need. |
Completion of confirmatory Phase III studies is still required. |
| Fast-Track Designation |
Granted to drugs intended to treat serious conditions and fill an unmet medical need. |
Completion of Phase III studies is still required although rolling submission is allowed, enabling portions of the NDA and data (clinical and non-clinical) to be submitted as it becomes available. |
| Breakthrough Therapy Designation |
Granted to drugs that may demonstrate substantial improvement over existing therapies in early clinical trials. |
Commercial application may be submitted based on early clinical evidence (completion of Phase III may not be required at time of initial submission). All benefits of the fasttrack designation (i.e., rolling submission) are automatically built in. A single cross disciplinary project manager is assigned at the FDA and commitment is made to frequent the FDA/ sponsor meetings throughout the development and review periods. |
| Priority Review Designation |
May be granted at the time of NDA submission to drugs which have achieved any of the above three criteria. |
Shortens the statutory review period from 10 months to six months for new chemical entities. Can also apply for a priority review in the case of supplemental applications in which case the review timeline is shortened from 10 months to six months. |
This article focuses on the US FDA Breakthrough Therapy Program and considers the impact of receiving a breakthrough designation early in clinical development and the challenges for accelerating CMC development activities to meet the expedited clinical development timelines.
Accelerated clinical and safety programs under the BT designation could lead to marketing applications up to two years or more earlier than a more conventional clinical development program. Review of potential CMC development programs required developing a formulation and manufacturing process capable of providing a sufficient reliable supply of product to patients at the time of approval on an indication designated as BT is likely to occur before all “traditional” CMC studies and data sets can be completed. This will require risk-based prioritization of time, resources and materials to accelerate certain activities and provide sufficient data and information to ensure an adequate supply of quality product for patients at the time of approval.
This article uses four case studies to exemplify how two different BT clinical development program scenarios could each impact a small molecule and a large molecule CMC development program. Using these scenarios, a range of CMC challenges are identified and proposals for progression discussed.
Introduction to Case Studies – Development Project Background
In this section, high-level development project plans are discussed examining the impact on CMC development of outcomes from clinical programs. From a drug development perspective and review within industry of current projects designated as breakthrough therapies, there are many scenarios. To simplify the discussion, two potential clinical development scenarios for the timing of receiving BT designation are discussed. These have been chosen when BT designation is given relatively early in the clinical program leading to greatest challenges for CMC development, where CMC is on the critical path. These two scenarios result when outstanding clinical findings for a serious disease or condition are observed from:
- Phase 1 studies in patients
- Phase 2 studies
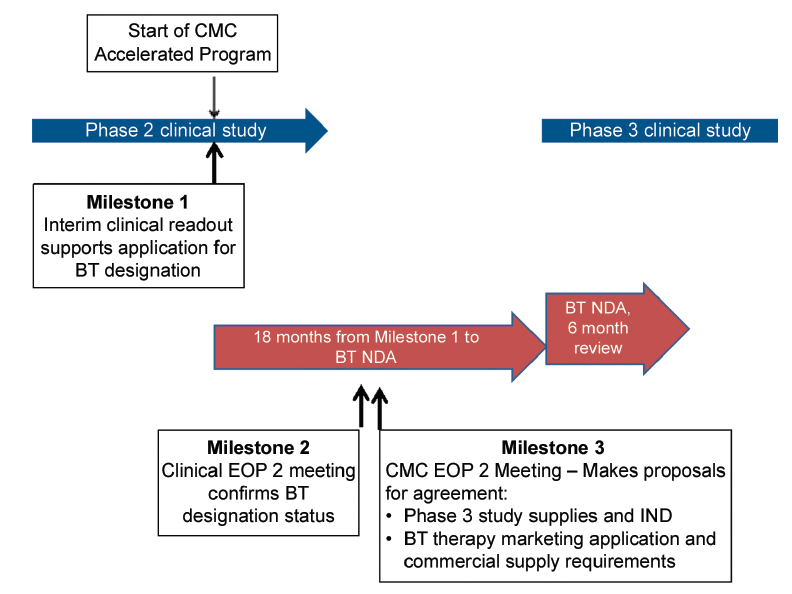
In Figure 1, a well-controlled study for a serious disease or condition leads to application for breakthrough therapy designation based on outstanding Phase 1 data. These studies would be conducted in patients, e.g., in oncology indications rather than volunteers and are unlikely to be comparative as required ideally by FDA guidance. At Milestone 1, outstanding clinical data are obtained leading to application for BT designation. It is possible that in the clinical End of Phase (EOP) 1 meeting, designation of BT status could be granted and a comparative Phase 2/3 clinical study agreed. Assuming good outcomes from the Phase 2/3 study, a marketing application could be filed. It is likely that clinical lifecycle expansion program (s) could commence at approximately the time of marketing application filing, which will be another challenge for the CMC development team in terms of product choice and supply.
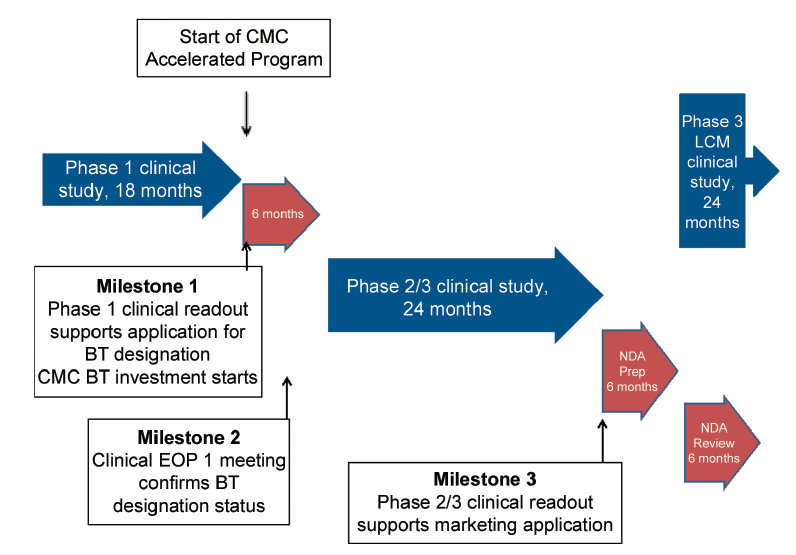
For this development scenario, it is assumed that there is approximately 36 months from receipt of outstanding Phase 1 clinical data (Milestone 1) and filing for a marketing application. For comparison, it could be expected that a “traditional” development program would be approximately 36 months from receipt of good Phase 2 clinical data to NDA filing.
In Figure 2, outstanding clinical data for a serious disease or condition emerge from an interim readout of a well-controlled Phase 2 study, which supports a company considering application for breakthrough therapy designation (Milestone 1). In this case, confirmation of outstanding clinical data would lead to a formal BT designation (Milestone 2) and this could lead to a marketing application filing six to nine months later based on these Phase 2 data. It is also likely that a confirmatory Phase 3 clinical study as well as clinical lifecycle studies could commence about the time of BT NDA filing. In this case, a Milestone 3 is given where the CMC team review CMC challenges with FDA.
In this case, there would be approximately 18 months from receipt of a good clinical signal to a marketing application filing.
CMC Case Studies and Topics for Discussion with FDA relating to Potential Flexibility
Four case studies are presented to highlight the variety of CMC issues that could be faced when clinical programs are accelerated and these are given as follows:
Case Study 1 – accelerated development of small molecule commercial formulation, non-ICH. stability data at time of marketing application and approval, and non-standard bioequivalence study.
Case Study 2 – accelerated development of a large molecule leading to non-ICH stability package and absence of PPQ data for drug substance and drug product in the marketing application.
Case Study 3 – accelerated development of a small molecule, which requires launch of clinical formulation from clinical manufacturing site and rapid change for patients to commercial formulation sourced from a commercial manufacturing site.
Case Study 4 – accelerated development of a large molecule, which for patients requires launch from clinical manufacturing site and rapid change to manufacture at a commercial site.
In all cases, proposals are made to optimize availability of patient-acceptable drug product to patients.
Common to all scenarios is the request to provide regulatory flexibility. Topics of interest are discussed with suggestions provided with justifications regarding approaches, which could be taken. In all discussions, it should be clear how the risks of different levels of information compared with a “traditional” application and the risks of supply of quality product to patients are mitigated. This risk mitigation strategy should be thoroughly explained and justified to the Agency.
There are other CMC issues, which could arise, for example setting of specification acceptance criteria from limited data, and changes of route of drug substance synthesis, which will allow facile provision of materials. These have not been exemplified in these case studies.
It should be noted that the working estimate for time saved in an accelerated clinical program is approximate 18 to 24 months. This translates to 18 to 24 months less time not only to complete all the activities necessary for CMC approval, but also to complete the activities necessary to launch and maintain commercial supply directly after approval.
To simplify the examples and focus on the main CMC issues, the following general assumptions are made regarding CMC development:
- There is no “front loading” of CMC development activities in the Phase 1 case studies. While “front loading” may be considered an expected business risk given the goal is the pursuit of unmet medical needs, there are large uncertainties regarding clinical outcome, which arises much later in the program. Large companies have multiple projects to fund, resource and hence prioritize, and small companies are unlikely to have the investor support.
- Some front loading is assumed for the Phase 2 case studies since it is assumed that a company will support its own judgment that it has a BT drug candidate, prior to FDA granting BT designation, i.e., between Milestones 1 and 2 in Figure 2.
- Small molecule drug substance synthesis is relatively straightforward.
- For small molecule, drug product is a tablet.
- For large molecules, a platform of monoclonal technology is used.
- Drug substance and drug product are stable.
Case Study 1 – Small Molecule, Phase 1 Entry
Assumptions for this case study in addition to the general assumptions given above are:
- “Simple” formulation used for Phase 1 studies – e.g., powder blend filled into a capsule
- BCS Class 1 drug substance (high solubility, high permeability)
- Limited time and availability of drug substance before Phase 2/3 study start to develop commercial formulation using QbD approach. A QbD approach will be used, however.
- Availability of sufficient drug substance for manufacture of clinical supplies is rate limiting and amount restricts commercial formulation development studies to small scale.
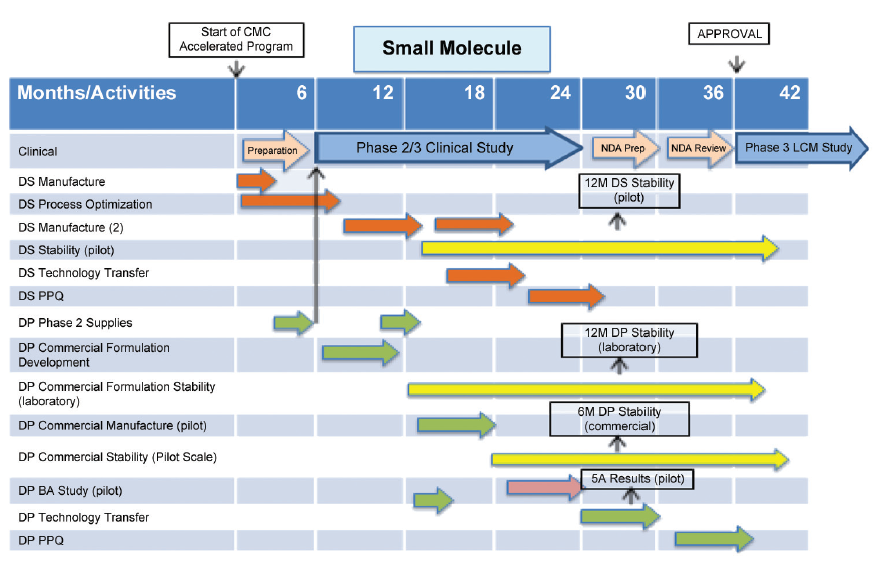
An outline CMC development plan is given in Figure 3 for a small molecule, which enters BT designation after Phase 1 clinical studies.
In the following CMC development plans, clinical programs are given in the top activity row, timescales and activities being taken from Figures 1 and 2. Drug substance development activities are given in orange, drug product activities in green and stability studies in yellow. CMC issues, which arise from this plan, are:
- Non robust formulation used to supply Phase 2/3 study due to lack of time and drug substance.
- Formulation change required for commercial supply. Bioavailability study conducted using commercial formulation manufactured at pilot scale.
- Reduced data set on commercial formulation, e.g., stability data.
- Process Performance Qualification (PPQ) of drug product conducted in a phased manner and completed post approval.
These challenges would be identified for discussion with the FDA early in the accelerated CMC development program, approximately in the range months three to six in Figure 3, when submitting the IND for the Phase 2/3 study.
Topics for Discussion with the FDA Relating to Potential Flexibility
Formulation Development and Bioequivalence – A robust formulation is required to supply patients bioequivalent to the formulation used in the pivotal Phase 2/3 clinical studies.
Proposal for Consideration – Present approach to formulation development, particularly the QbD approach to development of the commercial formulation as part of the IND Phase 2/3 submission along with the clinical strategy. Propose to demonstrate bioequivalence in two steps:
-
First demonstrating bioequivalent of commercial formulation at a 1/10th scale with Phase 2/3 formulation data in submission.
-
Followed by in vitro dissolution comparison of PPQ batches as given in FDA Guidance, Waiver of In Vivo Bioavailability and Bioequivalence Studies for Immediate- Release Solid Oral Dosage Forms Based on a Biopharmaceutics Classification System5 and SUPAC IR Guidance.6 Submit the results of the scale-up and characterization of the initial PQ batches in support of the CMC section with a commitment to complete PQ post approval in alignment with the FDA Guidance on the Process Validation Lifecycle.
Supporting Data – There is good assurance that the commercial scale drug product of this BCS Class 1 drug will be bioequivalent to the formulation used in Phase 2/3 studies. Bioequivalence data are provided in the NDA comparing pilot scale commercial formulation and Phase 2/3 formulation, and additional data are provided during review showing in vitro equivalence of commercial scale and pilot scale batches of commercial formulation. Given the benefit of providing product to patients at the time of BT approval, this approach is considered low risk.
Shelf Life, Drug Product – A shelf life of at least 18 months of shelf life is needed to maintain small molecule drug product in the supply chain for patient availability given the long lead-time for production and potentially low demand, at least initially.
Proposal for Consideration – Initiate the CMC section submission with 12 months data from three laboratory scale batches and six months data from three pilot scale batches of the commercial product packaged in the commercial pack assuming with a commitment to provide additional data during review and normal post approval commitments.
Supporting Data – Stress and accelerated studies during development demonstrate that the product is not prone to significant degradation or changes, confirmed by 12 months laboratory scale date. There are also data from stress and accelerated studies showing that there is no impact of scale on drug product chemical, physical and subjective stability. Assuming a rolling submission as proposed, further data could be provided during review, e.g., 18 month laboratory scale and nine and/ or 12 month pilot scale data. Commercial scale stability data would commence about the time of or shortly after projected approval to confirm this approach as low risk, even though it is different from ICH Q1A(R2) Stability Testing of New Drug Substances and Products.7
Phased Process Performance Qualification – At the time of anticipated clinical approval, the traditional process performance qualification of the large a molecule drug product will not be completed.
Proposal for Consideration – Supply product to patients immediately after approval with material from the first performance qualification batch.
Supporting Data – This approach is supported by the opportunity to use the concurrent release of PPQ btches approach given in FDA Process Validation guidance.8 Given the benefit to patients and the assurance that this batch and subsequent batches comply with the PPQ protocol there is good assurance that quality product will be provided to patients. Additionally, it could be suggested that the PPQ protocol is provided to reviewers during review. Overall, this approach is considered low risk.
- 5FDA Guidance for Industry: Waiver of In Vivo Bioavailability and Bioequivalence Studies for Immediate-Release Solid Oral Dosage Forms Based on a iopharmaceutics Classification System, August 2000, U.S. Food and Drug Administration (FDA), www.fda.gov.
- 6Immediate Release Solid Oral Dosage Forms: Scale Up and Post-Approval Changes: Chemistry, Manufacturing and Controls, In Vitro Dissolution Testing, and In Vivo
- 7Bioequivalence Documentation (1995) and SUPAC-IR Questions and Answers about SUPAC-IR Guidance, 1997.
- 8ICH Q1A(R2) – Stability Testing of New Drug Substances and Products, Step 4, 6 February 2003, www.ich.org.
Case Study 2 – Large Molecule, Phase 1 Entry
Assumptions for this case study in addition to the general assumptions given above are:
- Solution formulation for Phase 1.
- Limited time to develop and scale-up drug substance process prior to start of Phase 2/3 clinical studies.
- Studies to optimize and scale-up the drug substance process focus on process reliability over yield and cost of goods.
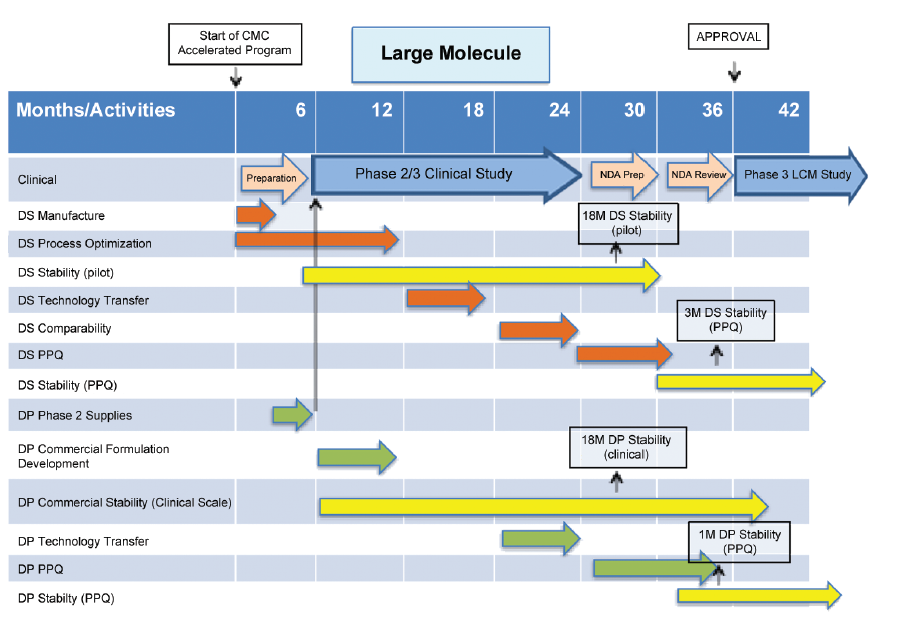
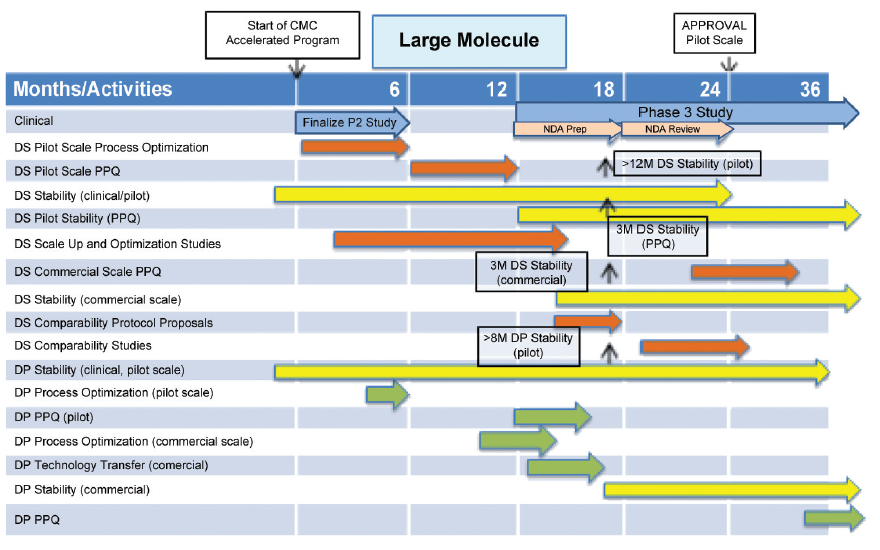
An outline CMC development plan is given in Figure 4 for a large molecule, which enters BT designation after Phase 1 clinical studies. CMC issues, which arise from this plan, are:
- Stability data for drug substance and drug product do not comply with ICH Q5C, Stability Testing of Biotechnological/ Biological Products9 at time of proposed filing of marketing application.
- Process Performance Qualification (PPQ) of drug substance and drug product are not complete at time of proposed filing of the marketing application. It is conducted in a phased manner and completed post approval.
- Patients are proposed to be supplied from PPQ batches.
These challenges would be identified for discussion with FDA early in the accelerated CMC development program, approximately in the range months three to six in Figure 4, when submitting the IND for the Phase 2/3 study.
Topics for Discussion with the FDA Relating to Potential Flexibility
Storage Period, Drug Substance – A storage period of at least 12 months is required for the large molecule drug substance to provide sufficient time to allow compounding into multiple batches of drug product without waste. Shorter storage periods would lead to unacceptable levels of waste from the batch size of drug substance justified and proposed in the NDA, or increased resources conducting re-testing.
Proposal for Consideration – A storage period for commercial scale drug substance of 12 months is proposed based on provision of three month data from one batch of commercial scale biologic drug substance provided in BLA with a commitment to provide six months during review and to supply further commercial scale data to a pre-agreed protocol.
Supporting Data – This proposal is supported by 18 months good data on pilot/ clinical batches with the option to provide additional data (e.g., 24 months) during review. There is additional assurance regarding absence of stability differences due to scale from other earlier studies with similar monoclonal antibodies manufactured using the same platform technology, and evidence supports that it is a stable drug substance. Given the good stability performance of this drug substance, this is considered a low risk approach even though it is different from ICH Q5C, Stability Testing of Biotechnological/Biological Products.9
Shelf Life, Drug Product – A shelf life of at least 18 months is needed to maintain product in the supply chain for patient availability given the long lead-time for production and potentially low demand, at least initially.
Proposal for Consideration – An initial expiry 18 months is proposed based on 18 months real time data provided in the commercial pack from two pilot (clinical) scale batches for a stable drug product with the option to provide 24 months data during review. Commercial scale stability studies are performed to a pre-agreed protocol. One month data from a PPQ batch could be available late during the review period. The protocol for commercial stability studies is proposed to be a matrix of time period, test and size of container.
Supporting Data – This is supported by results from extensive development studies (e.g., accelerated and stress studies examining as independent variables, scale of manufacture and volume size of the commercial pack) showing that scale (essentially time of filling) of an aseptically-filled solution drug product and volume size of commercial pack do not impact stability. Stability data from at least one technology transfer batch (three months) at commercial scale could be available at time of filing of BLA. Given the good stability performance of drug product and the sponsor’s commitment to comply with the protocol this is considered a low risk approach even though it is different from ICH Q5C, Stability Testing of Biotechnological/ Biological Products.9
Additional note – ISPE sees an opportunity to provide guidance/best practices related to designing and documenting early stability programs to support expiration dating for product launch.
Process Performance Qualification – Clinical approval overlaps with the execution of the process performance qualification studies for drug substance and drug product, which for a large molecule marketing application results are required in BLA submission. The product has a long lead-time in production. The production timing is such that to have material available for launch supplies as soon as possible following clinical approval, it will be necessary to utilize the PPQ batches for commercial supply.
Proposals for Consideration – Two proposals related to provision of process performance qualification information are:
-
Submit the available scale-up, comparability and characterization data along with the PPQ protocol with a commitment to provide the data as it becomes available during review and, depending on timing, concurrent with product release. This assumes data meet all requirements under the protocol.
-
Submit drug substance comparability protocol and data, and PPQ protocols for drug substance and drug product in the BLA. Provide all data for drug substance, which should be completed, during review. For drug product, provide all available data during review.
-
Drug product is proposed for supply to patients using material from PPQ batches complying with PPP protocol criteria
Supporting Information – Considering the compatibility has already been established, providing PPQ data in a phased manner as suggested above provides maximum information for review prior to approval. Process performance qualification contains many repeat studies conducted as part of the comparability program and hence the risk of failure to comply with PPQ protocol criteria is low.
Given that platform technology is being used, that comparability studies are completed and acceptable comparing commercial scale batches with clinical batches and a good package of drug substance and drug product information is included in the BLA to support presentation of the drug product process performance qualification protocol, it is proposed that drug product from process performance qualification batches complying with the protocol is used to supply patients. This suggestion is in line with the concurrent release of PPQ batch approach given in FDA Process Validation Guidance.4 It is proposed that PPQ qualification report is provided to the Agency on completion, post approval if required. This is considered a low risk approach.
Case Study 3 – Small Molecule, Phase 2 Entry
Assumptions for this case study in addition to the general assumptions given above are: Phase 2 formulation is fit-for-purpose, however, not optimized in terms of robustness or commercial presentation (Phase 2 studies are blinded).
- Strong need for patients to introduce “improved” formulation as soon as possible.
- Drug substance route is adequate for Phase 3 clinical and toxicology study supply.
- BCS Class 1, high solubility, high permeability.
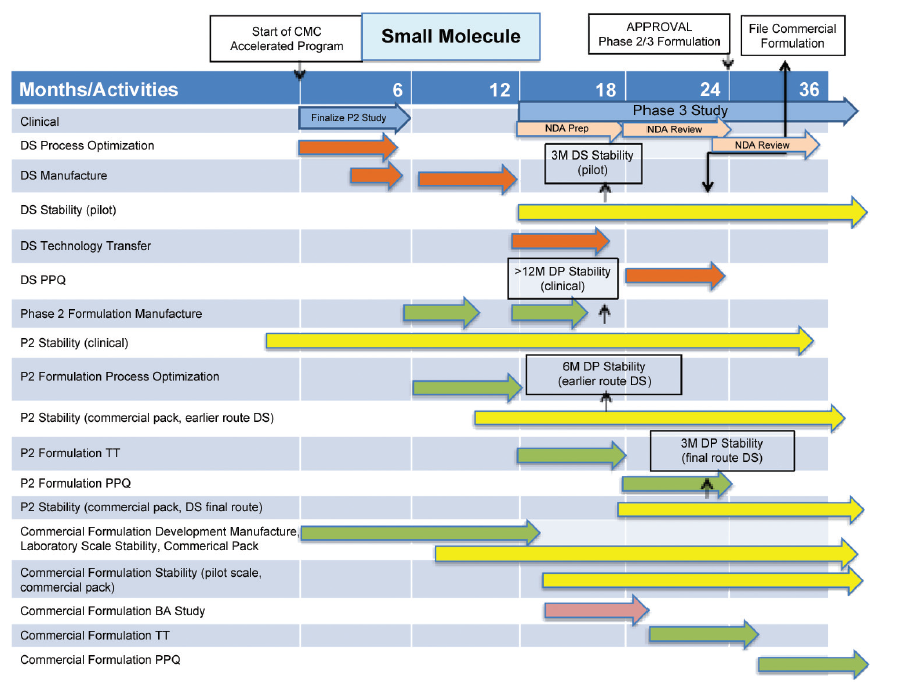
An outline CMC development plan is given in Figure 5 for a small molecule, which enters BT designation after Phase 2 clinical studies.
CMC issues, which arise from this plan, are:
- Propose launch of “fit-for-purpose” Phase 2 formulation from pilot/clinical manufacturing site.
- Reduced stability dataset for Phase 2 formulation from pilot/clinical site at time of marketing application.
- File for change of formulation and site of manufacture approval before approval of Phase 2 formulation.
- Reduced stability dataset for commercial formulation from commercial manufacturing site at time of marketing application.
These CMC issues would be evident at the start of an accelerated program and hence could be discussed with FDA at or shortly after Milestone 2 in Figure 2 when there is agreement to BT designation.
Topics for Discussion with the FDA Relating to Potential Flexibility
Formulation and Site of Manufacture Change – The clinical formulation and site of manufacture are not suitable for long term supply to patients.
Proposal for Consideration – The clinical formulation is fit-for-purpose for Phase 2 clinical studies, which are blinded. It is not viable for long term commercial supply to patients. In order to meet patient needs at the time of proposed clinical approval it is proposed to supply Phase 2 clinical formulation sourced from the clinical manufacturing site. Change to an “improved” formulation to meet patient needs better and to source from a commercial manufacturing site is proposed with the marketing application submitted before approval of the Phase 2 formulation.
Supporting Information – Feedback from patients and medical practitioners during the Phase 2 is that the tablet dosage form is too large. Given the patient population intended to be treated there is a strong requirement to re-formulate to a smaller dosage form more acceptable to patients. At the time of anticipated clinical approval the commercial formulation cannot be developed and shown bioequivalent to the Phase 2 to allow initial launch of the commercial formulation.
Non-Standard Stability Package for Phase 2 Formulation Marketing Application – A shelf life of at least 18 months is needed to maintain product in the supply chain for patient availability given the long lead-time for production and potentially low demand, at least initially. A retest date of at least 12 months is required to support smooth progression of drug substance into drug product. At the time of marketing application for the Phase 2 formulation manufactured at the clinical manufacturing site the stability package does not comply with ICH Q1A(R2).
Proposal for Consideration – Stability data at time of filing of Phase 2 formulation marketing application:
- 3 month of 3 batches of drug substance manufactured at pilot scale using commercial synthetic route supported by 12 months from >1 batch manufactured by an earlier synthetic route
- >12 month from 1 batch of drug product packaged in the clinical pack
- 6 months from 3 batches of drug product manufactured from an earlier synthetic route packaged in commercial pack
- 3 months from 3 batches of drug product manufactured using final route synthesis drug substance and packaged in commercial packs available during review
Supporting Information – A shelf life of 18 months is proposed for the Phase 2 formulation packaged in the commercial pack based on greater than 12 months data for this formulation packed in the clinical pack plus three months data using final synthetic route drug substance and six months data from the previous route. Accelerated data on both drug product and drug substance show no difference in stability due to route of synthesis of drug substance. Development studies also show no difference in stability between drug product assembled into clinical and commercial packs. The Agency will be informed immediately if data are generated outside an agreed protocol. Given the substantial amount of data for this stable drug product the proposed shelf life and overall approach is considered low risk even though it is different from ICH Q1A(R2) Stability Testing of New Drug Substances and Products.8
A retest date of 12 months is proposed for drug substance supported by at least 12 months satisfactory data from an earlier synthetic route and three months of accelerated data for a stable drug substance showing good stability from three batches of drug substance manufactured at pilot scale. For a stable drug substance, the proposed retest date is considered low risk even though it is different from ICH Q1A(R2) Stability Testing of New Drug Substances and Products.8
File Marketing Application for New Formulation Before Phase 2 Formulation Approved – It is proposed to file the new formulation and site based on the information given above during review of Phase 2 formulation (for example about one month before expected approval) with the proposal that review of this application is also subject to BT timelines. The benefit would be a more reliable supply of quality product to patients.
Proposal for Consideration – This non-standard regulatory process would require much discussion and prior agreement from the agency. There are many points of discussion, however, to take stability and bioavailability of drug product as an example, the following data should be available at time of filing for commercial formulation marketing application:
- BA study comparing new formulation using material from pilot scale manufacture vs Phase 2 formulation
- 3 month stability data from pilot scale batches of drug product with 6 month data available during review of new formulation
Supporting Information – It is proposed to demonstrate bioequivalence in two steps:
- First demonstrating bioequivalent of commercial formulation at a 1/10th scale with Phase 3 formulation with data in submission.
- Followed by acceptable in vitro dissolution comparison of PPQ batches as given in FDA Guidance, Waiver of In Vivo Bioavailability and Bioequivalence Studies for Immediate-Release Solid Oral Dosage Forms Based on a Biopharmaceutics Classification System5 and SUPAC IR Guidance.6
- An 18 month shelf life for drug product would be justified by:
- Extensive development data from pre-formulation studies
- Stress and accelerated data comparing the commercial formulation and the Phase 2 formulation
- Stress and accelerated data comparing different scales of manufacture of the commercial formulation
- Real time data on pilot scale formulation
- For a stable drug substance, the proposed retest date is considered low risk even though it is different from ICH Q1A(R2) Stability Testing of New Drug Substances And Products.7 It is a stability package which is similar to an ANDA application particularly if one or three months stability from at least one commercial scale batch of drug product were provided during review.
Case Study 4 – Large Molecule, Phase 2 Entry
Assumptions for this case study in addition to the general assumptions given above are:
- Phase 2 solution formula proposed as commercial dosage for filing
- Focus time available to filing on process optimization studies
- Immediate scale up required to supply anticipated commercial demand
An outline CMC development plan is given in Figure 6 for a large molecule, which enters BT designation after Phase 2 clinical studies. To meet anticipated clinical approval timelines it is proposed to launch from pilot scale drug substance and drug product sites, which obviously assume that projected initial market demand can be met; however, projected demand requires scale up as soon as possible. Delay of approval of the commercial manufacturing site may result in stock outs and rationing of supplies to patients.
Given the patient need and availability of expertise and resource to support drug substance process development and scale-up, there needs to be a balance of resource applied to qualification studies for pilot scale manufacture and scaleup studies and qualification of production scale manufacture.
These CMC issues would be evident at the start of an accelerated program and hence could be discussed with the FDA at or shortly after Milestone 2 in Figure 2 when there is agreement to BT designation
Topics for Discussion with the FDA Relating to Potential Flexibility
Site of Manufacture Change, Balance of Pilot Scale PPQ Studies, and Commercial Scale-Up and PPQ Studies – This CMC scenario is considerably different from a “traditional” submission for a BLA and would require substantial discussion with the Agency, an example of major differences from the ‘traditional’ approach being: Filing with limited qualification data from pilot scale.
Proposal for Consideration – Protocol and study design for PPQ studies to support a BLA application for pilot scale manufacture requires discussion and agreement with the Agency, potentially on more than one occasion. During these discussions, it would be appropriate to agree studies to support PPQ of commercial scale manufacture and the timing of the proposed marketing application for commercial scale manufacture. The date of filing for commercial scale manufacturing is not given in Figure 6; however, from a patient viewpoint, this should be as early as possible.
Supporting Information – Information for the discussion would be developed using a risk-based approach utilizing prior knowledge from the platform technology, and development studies performed for this drug substance. Studies for PPQ at commercial scale would be informed by parallel studies conducting PPQ of pilot scale manufacture
Shelf Life Request – The amount of drug substance stability data at pilot scale should be sufficient, for example greater than 12 months on an early batch, and six months from three batches from proposed initial scale of supply. The amount of stability data for pilot scale drug product should also be sufficient given that there will be at least six months data on three batches from the intended initial scale of supply. A shelf life of greater than six months would be requested to maintain supplies to patients, for example 18 months (re-labeling after approval of shelf life extensions is not practical).
Conclusion
If a development project generates outstanding clinical data for a serious disease or condition, it is likely that a company or the FDA will request that formal application is made for BT designation. If the development project team considers this a good possibility, the implications on CMC development are significant. For example:
- BT nomination could give insufficient time to complete all ‘traditional’ CMC studies.
- BT CMC work for filing should use a risk-based approach to prioritize time, resources and materials to provide data and information to support a BT NDA filing, and to ensure supply of quality product to patients.
- Given the assumption that CMC is not complete, there is likely to be more post approval activity, for example:
- More stability data
- Additional confirmatory validation (process robustness) studies
- hanges of site, scale of manufacture, raw material supplier - Changes of drug substance synthetic route
- Change of formulation with supporting bioavailability studies
In conclusion, there is sufficient justification in all the above cases studies to discuss with the Agency filing using more flexible regulatory approaches to provide patients with an exciting new drug based on providing assurance of quality.
Dialogue with the FDA should be early, fast and effective to provide the CMC development team with answers to which they can respond in the limited time available to ensure that, given approval, patients can be supplied with quality drug product. It is submitted that the current process of providing a briefing document about three months before a meeting with the FDA and receiving formal answers about a month following the meeting is an insufficient level of assurance that answers can be addressed so that there is a positive impact on a NDA/BLA filing strategy. Better interactions with the FDA are being employed to facilitate accelerated approvals, for example use of:
- “Informal” telephone conversations. There still the responsibility of the sponsor to record the conclusions of the conversation.
- IND amendments
BT designation produces many CMC challenges which a sponsor and the FDA need to address using a risk-based approach to assure sufficient information available to support approval and supply of quality product for serious disease or condition’ demonstrating “substantial improvement over existing therapies to patients.