Biopharmaceutical Manufacturing Process Validation and Quality Risk Management

This article was published in the May/June 2016 edition of Pharmaceutical Engineering® magazine. It is one of five articles nominated for the Roger F. Sherwood Article of the Year Award, all which will be posted to iSpeak throughout the week of 5 December.
25 Process validation today is a continual, risk-based, quality-focused exercise that encompasses the entire product life cycle.
Manufacturing processes for biopharmaceuticals must be designed to produce products that have consistent quality attributes. This entails removing impurities and contaminants that include endotoxins, viruses, cell membranes, nucleic acids, proteins, culture media components, process chemicals, and ligands leached from chromatography media, as well as product modifications, aggregates, and inactive forms. Manufacturing processes should be validated by applying a scientifically rigorous and well-documented exercise demonstrating that the process, and every piece of equipment used in it, consistently performs as intended, and that the process, when operated within established limits, generates a product that routinely and reliably meets its required quality standards. The principles of process validation were initially established in the 1987 US Food and Drug Administration (FDA) document “Guideline on General Principles of Process Validation,” which defined process validation as “establishing documented evidence which provides a high degree of assurance that a specific process will consistently produce a product meeting its pre-determined specifications and quality attributes.”1 This definition has since been adopted in guidance documents worldwide, including the current good manufacturing practices (cGMP) regulations promulgated by European regulatory agencies and the International Conference on Harmonisation (ICH). When the 1987 FDA guidance was published, validation during early stages of product development (before Phase 1 clinical trials) was minimal:
- Qualifying master and working cell banks
- Demonstrating adequate virus clearance (removal and inactivation) by the manufacturing process
- Validating sterilization and aseptic processes used to manufacture the drug product
At that time, most process validation activities were conducted in the later stages of product development, primarily during Phase 3 clinical trials, in preparation for filing a biologics license application (BLA) and eventual commercialization of the product. These activities included:
- Identifying critical process parameters (CPPs): those independent process inputs or variables related to each individual unit operation in a manufacturing process that directly affected product quality
- Conducting range studies on these parameters to determine the points at which the process fails to yield acceptable product
- Producing a series (three to five) of consecutive full-scale conformance lots in qualified equipment under cGMP conditions
Equipment qualification involved confirming and documenting that the design, installation qualification (IQ), operation qualification (OQ), and performance qualification (PQ) of the manufacturing equipment were capable of satisfying the process requirements. Analytical methods used for in-process testing and final product release were validated prior to initiation of full-scale conformance lots. After conformance lot approval, the validated process could not be materially modified without revalidation to confirm that the process was still under control and still resulted in a product of acceptable (comparable) quality.
Evolution
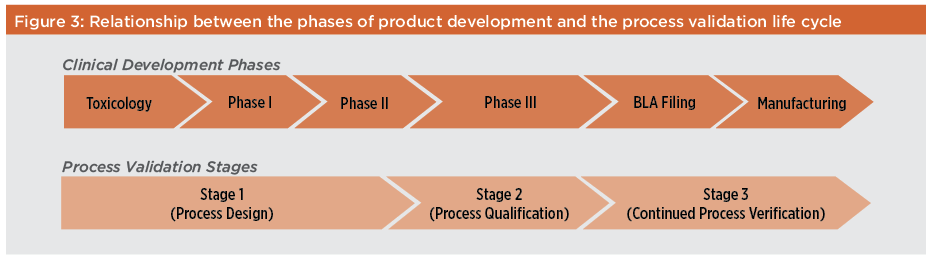
Since 1987 the concepts of validation in general, and process validation in particular, have evolved. Process validation is now viewed as a continuum of activities rather than a series of discrete actions that are performed once and rarely repeated. Regulatory authorities also now consider process validation as encompassing not only a full demonstration of process consistency and understanding, but also ongoing verification to ensure the process remains within its qualified design space and product consistently meets all specifications. In addition, regulatory authorities expect companies to develop unique validation protocols suited to their individual organizations. These protocols are no longer based on conformance to a fixed set of guidelines, but are designed using a risk-based approach that identifies and controls potential risks within the manufacturing process. This approach to overall product development and validation was outlined in 2004 by FDA in “Pharmaceutical cGMPs for the 21st Century—A Risk-Based Approach,”2 and reinforced in 2005 with the approval of ICH Q9,3 which formalized the requirements of quality risk management for the pharmaceutical industry. This was further defined in FDA’s 2011 guidance on process validation4 and the European Medicines Agency (EMA) 2012 draft process validation guideline.5 As a result, validation must now take a continual life cycle approach. This shift acknowledges the need for improvement in manufacturing processes, in alignment with the quality by design (QbD) approach.6 In addition, both FDA and EMA guidelines are now in line with ICH Q8(R2), Q9, and Q10, and Q11; both also require adherence to cGMP regulations. But there are subtle differences between the two. FDA’s 2011 guidance divides the validation of a manufacturing process across the life cycle of the product into three stages: process design, process performance qualification, and continued process verification. The EMA guideline does not divide process validation into stages. It also allows for a hybrid approach that combines the new process validation guidance with the traditional approach; FDA requires that the new guidelines supersede the traditional practice.
Quality Risk Management
To meet the regulatory requirement that commercial pharmaceutical manufacturing processes be “validated with a high degree of assurance,”7 regulatory authorities now consider a systematic risk analysis and management program to be a critical component of validation.8 A quality risk management program (see Figure 1) will encompass risk control, risk review, and, most importantly, risk assessment, which is the most critical aspect for process validation.
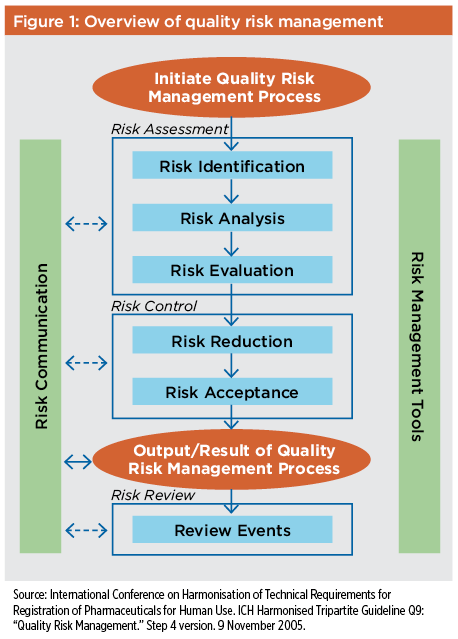
Risk assessments should be based on sound science, process characterization information, and data collected from both scaled-down models of the manufacturing process and actual product batches produced during clinical development and scale-up. The data should include information about the source and quality of all materials used in the manufacturing process, as well as the effect of each material or procedure used in the process on the quality, efficacy, and safety of the final product. Risk assessments should be conducted throughout the product life cycle, starting with process design and continuing through ongoing assessment of commercial manufacturing operations. Risk assessment approaches used initially to determine product critical quality attributes (CQAs) include risk ranking and preliminary hazard analysis (PHA). These are illustrated in a 2009 case study for a monoclonal antibody bioprocess development, which is a practical guide on how to use both QbD and life cycle approach to validation.9 Later risk assessments include process risk assessment (PRA), which is conducted using failure modes effects analysis (FMEA); failure modes effects criticality analysis (FMECA); or the hazard analysis and critical control point (HACCP) methodology. Risk assessments should be conducted at phase-appropriate intervals, and any time that changes are made to the manufacturing process. Depending on situation and need, they can, and should be, both formal and informal. As the product matures and additional process knowledge accrues, risk assessment and analysis will become more comprehensive, helping to determine the potential effects of even subtle manufacturing process changes on product quality.
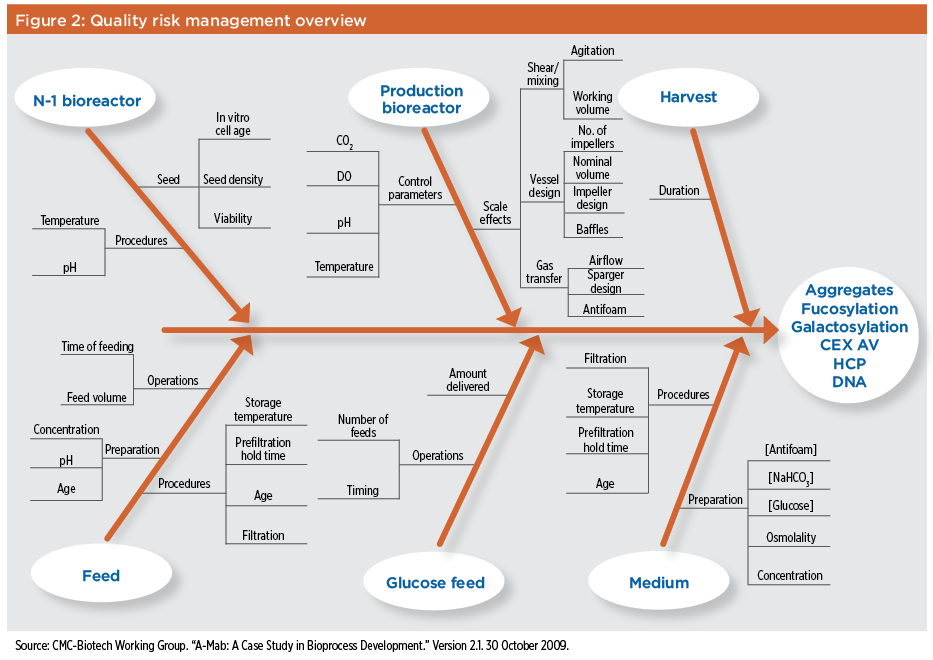
The glycosylation of recombinant proteins, for example, can be altered by a range of factors associated with cellular metabolism and metabolic flux as well as the efficiency of the glycosylation process. Since changes in glycosylation can have a significant effect on biopharmaceutical product pharmacokinetics, efficacy, and immunogenicity, it’s important to assess the risk of variations in the production bioreactor operating parameters and any possible effects on product glycosylation.10 This is especially important since subtle variations of nominally identical bioreactor operating parameters can alter glycosylation.11 It may be difficult to determine the effect of certain manufacturing parameters on glycosylation early in the product life cycle, however, due to the limited number of batches produced during clinical development and the limited clinical data available at that time. The potential risks associated with raw materials, process equipment, and manufacturing processes on biopharmaceutical product quality should also be part of the evaluation. The criticality of these risks should be determined, as should methods or policies designed to eliminate, mitigate, or control them. A quality risk management program will define and prioritize the operating parameters that must be controlled during a manufacturing process. In alignment with QbD, quality risk management acknowledges that it is not possible to achieve control of product quality by final product testing alone. Product’s CQAs should also be identified using appropriate risk assessments, and confirmed during process development and early-stage manufacturing. These CQAs should then be maintained throughout the product life cycle by carefully controlling and monitoring those CPPs that may affect them. By establishing the CQAs for a product, defining the acceptable ranges for each CPP to achieve these CQAs, and controlling those CPPs during manufacturing, it’s possible to define a design space for each process step that incorporates the acceptable operating ranges of all CPPs. This approach allows a manufacturing process to be optimized or changed as long as design space parameters are maintained. Staying within the process design space will eliminate the requirement for revalidation of the manufacturing process, encourage innovation, and allow process changes to be implemented with minimum regulatory delay and expense. An additional useful tool in conducting an initial risk assessment is the Ishikawa or fishbone diagram, which can be used to identify all possible causes for a given effect. Such an analysis is helpful, for example, in evaluating how different process parameters might affect certain process attributes. In the A-Mab case study mentioned earlier,9 a fishbone diagram was used to identify equipment design, control parameters, processing conditions, and starting materials for a production bioreactor and its seed reactor that might have posed a significant risk to the quality attributes of a monoclonal antibody product. This analysis, shown in Figure 2, helped assess the potential effect of each process parameter on product yield and cell viability of the culture. It also identified soluble aggregates, variability in glycosylation, deamidation, and levels of host cell protein or DNA at harvest.
Risk Assessment Tools
ICH Q9 recommends the use of such standard risk analysis tools as FMEA/FMECA and HACCP to quantify the risk associated with each step in a manufacturing process and determine CPPs.3 Additionally, risk ranking and PHA can be used to determine the CQAs.9 Individual risk assessment techniques are best used in a complementary manner to eliminate knowledge gaps. Before initiating any risk assessment the scope must be defined, the risk assessment tool chosen, an appropriate team selected, and any potential decisions that will be based on the assessment clearly stated. Defining the scope of the risk assessment will also help determine the proper team composition. Risk assessment teams should include all individuals required to bring the necessary expertise to the assessment; they may include representatives from validation, process development, quality, and manufacturing.8 A simple but effective approach to risk analysis is provided by Katz and Campbell:12 A manufacturing process is broken down to its constituent unit operations and the specific parameters of each operation are analyzed to determine whether that parameter poses a risk to product identity, strength, quality, purity, or potency. Since each unit operation intended is to meet or maintain some section(s) of the quality target product profile, identifying and managing those process parameters that affect the product’s CQAs constitutes the control strategy for that particular unit operation.
PHA
This risk assessment tool can be used to rank quality attributes based on the probability and severity of failure by leveraging prior knowledge to identify future risks to the patient.3 PHA produces a severity score, which considers risks to safety and/or efficacy based on prior knowledge elements. PHA also calculates a probability score based on the chances of a quality attribute affecting safety and/or efficacy by going outside of the currently established ranges.9 The probability and severity scores are multiplied to calculate the risk priority number (RPN), which allows the quality attributes to be ranked.
FMEA
FMEA is a methodology for identifying potential failure modes for a product or process; it is designed to assess the risk associated with those failure modes and to classify the severity of failures on the product or process. FMEA analysis ranks potential failure modes and identifies corrective actions to address the most serious concerns. Failure modes are defined as any errors or defects in a process or product, especially those that affect the product’s safety or efficacy. FMEA considers three factors in evaluating the effect of a failure:13
- Severity: the impact of failure
- Probability: the likelihood of failure
- Detection: the detectability of failure
These factors are assigned scores determined by the scale assigned for each one. The scores are multiplied to calculate the RPN, which ranks the failure mode, prioritizes risks, and evaluates risk mitigation.3 FMEA is best suited for the evaluation of equipment and manufacturing processes. FMEA/FMECA may be used in the PRA to identify parameters for screening in-process characterization studies.
HACCP
This systematic preventive approach to product safety addresses hazard identification, evaluation, and control rather than finished product inspection. Used for years in the food industry,14 HACCP can be applied to biopharmaceutical product development and manufacturing as a means of identifying the points in a process at which specified critical control points may be controlled, the limits of control available, monitoring requirements, and required corrective actions. For most biopharmaceutical product manufacturing processes, FMEA is generally used to determine risks associated with the manufacturing process. For those manufacturing processes where controlling hazards is a critical issue, however, HACCP may be more appropriate. This is because HACCP focuses on critical control points to prevent or eliminate hazards and risk, while FMEA focuses on the potential effects of any identified failure mode. An HACCP analysis, for example, may be better suited than an FMEA analysis for determining risks when a filling process for a biopharmaceutical molecule conjugated to a toxic compound relies heavily on environmental and manufacturing controls to ensure not just product quality, but patient and operator safety.
Risk Ranking
Risk ranking is used to assess product quality attributes and determine which must be controlled as CQAs. Risk ranking evaluates quality attributes based on their potential to affect the patient adversely multiplied by the level of confidence in the knowledge used to determine that effect. This is scored by evaluating known or potential effects on safety and/or efficacy.9 The uncertainty is scored by leveraging prior knowledge elements as recommended by ICH Q9. Scoring for each category should be established using a numerical system commensurate with the criteria for each category. The numerical scale used is considered arbitrary, provided it gives appropriate to the impact score. Risk ranking does not take into consideration the detectability or controllability of a failure; as a result, the criticality score will not change as product and process knowledge evolve. It will change, however, as understanding of the product increases. Risk ranking should be used during the initial assessment of product quality attributes and reevaluated over the course of the product life cycle at phase-appropriate intervals. An example of the type of risk analysis and ranking that can be used to assess the impact of raw materials or process parameters on product quality attributes and the assignment of CQAs is provided by Boychyn and Hart, who applied this approach in assessing the risk of adventitious agent contamination of raw materials used in cell culture media.15 Their assessment concluded that the highest risk for viral contamination in media was associated with use of raw materials containing animal-derived ingredients, materials that are a potential food for rodents, materials that are not highly purified, or when raw materials represented greater than 10% of the volume of the media. These factors had a risk potential several orders of magnitude greater than the next-highest set of raw material risks evaluated. As a result of this analysis, cell culture media containing the highest-risk raw materials should be subjected to viral inactivation processes before they are used in product manufacturing. A similar analysis by Kiss concluded that the highest-impact risk mitigation strategy was to provide an efficacious virus barrier at the point of use in the manufacturing facility.16
“Life Cycle” Process Validation
With the introduction of QbD and quality risk management, process validation has evolved from a traditional “fixed-point” manufacturing process following process validation to a “life cycle” methodology that enables more continuous improvement of manufacturing processes. In this modernized approach, manufacturing processes are continually reviewed during routine manufacture to ensure that adverse trends are identified and corrected before the product fails to meet its final specifications. These new process validation guidelines promote designing quality into the product rather than simply testing for quality in the finished product. As defined in the FDA January 2011 guidance, the life cycle approach specifies that traditional process validation, which typically relies on three consecutive successful full-scale conformance runs, should be replaced by a deliberate design process, commercial process qualification, and ongoing review of processes with increased use of continuous process monitoring.4 The relationship between the various phases of clinical development and commercialization of a biopharmaceutical product and the three stages of process validation (process design, process qualification, and process verification) is shown in Figure 3. As knowledge about the safety and efficacy of a product increases during its clinical development, so too does the knowledge of its manufacturing process. Now the CQAs of the product and CPPs of the manufacturing process, initially defined during process validation Stages 1 and 2, are continuously monitored and verified during Stage 3. This requirement for continued process verification remains throughout the commercial life of the product.
Stage 1: Process Design
During process design, the manufacturing process is developed, characterized, and then scaled up to commercial levels as outlined earlier in this paper. During Stage 1, product CQAs should be identified and the critical and key process parameters for the manufacturing process defined.17 Since CPPs must be maintained or controlled within their specified ranges to demonstrate process robustness and suitability, acceptable operating ranges for these parameters should be established during this stage. As described below, much process design and process development work can be done using scaled-down process models and high-throughput development techniques. FDA guidance recommends using statistical design of experiments to study the interaction of different process parameters using multivariate experiments.4 Process design during Stage 1 encompasses laboratory activities for process development and process characterization, as well as establishment of a commercial process control strategy. Key prerequisites include sufficient product characterization data to establish product CQAs, and sufficient scale-up/scale-down data to ensure that the laboratory models used in process characterization represent full-scale manufacturing performance. During Stage 1, a standardized approach such as that outlined in Figure 4 allows all unit operations, analytical methods, and product specifications to be scrutinized carefully and developed properly. Each CPP in the manufacturing process should also be classified.
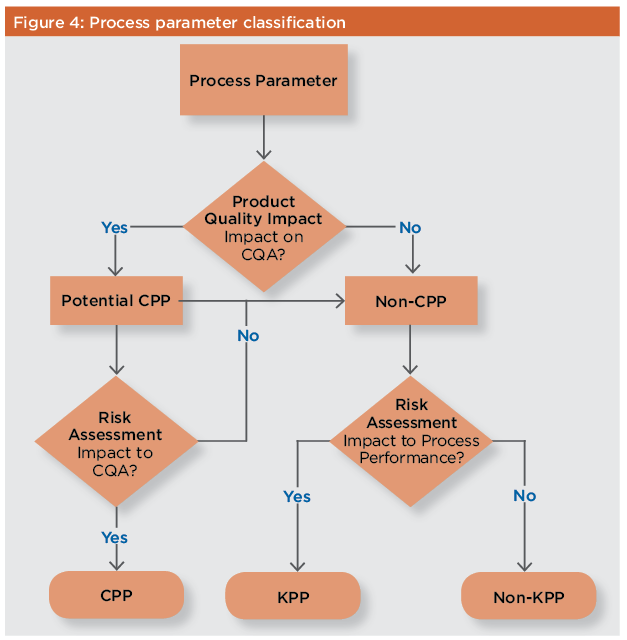
A CPP is “a process parameter whose variability has an impact on a critical quality attribute and therefore should be monitored or controlled to ensure the process produces the desired product quality.”18 Process parameters are classified as either critical or non-critical through risk assessment, as discussed above. These additional classifications, while not an absolute regulatory requirement, can be helpful during routine manufacturing to determine acceptable responses to process deviations or excursions. Non-CPPs may be divided into two discrete categories, key and non-key process parameters, in accordance with the definitions established by the Parenteral Drug Association. Non-CPPs that do not affect product quality, but may affect process performance such as yield, are classified as key process parameters (KPPs). Non-key process parameters (non-KPPs) are those that have no effect on process performance or product quality. CPPs, KPPs, and non-KPPs do not represent a continuum of criticality. While designation of a process parameter as CPP or non-CPP is based on a continuum of risk, the decision to classify parameters as KPP or non-KPP is binary.19 There is no universal definition for categorization of process parameters as CPP or non-CPP, and as such these categorizations are not necessarily recognized by global regulatory authorities.19 –20 Regulatory authorities generally discourage the use of key and non-key parameters in regulatory submissions. However, it is possible to define categories of process parameter criticality to meet individual program requirements. The A-Mab case study provides an example of how criticality rankings can be customized.
In this study, critical process parameters were classified as either CPP or well-controlled CPP (WC-CPP). Non-critical process parameters were designated non-CPP or general process parameter (GPP). This process acknowledges that although criticality assignment is binary, the potential effect of a process parameter can depend on a variety of factors, including the controllability of an individual process parameter.9 A criticality assignment process with greater granularity can facilitate better decisions regarding controls for process parameters. A list of activities typically performed during process design is provided in Table A along with the deliverable used to document completion of the activity and its outcome.

Careful planning and forward thinking during Stage 1 are essential to a successful validation program. The life cycle validation approach requires a strong foundation as quality must be built in from the start. Good studies in Stage 1 strongly contribute to Stage 2, process qualifications.
Stage 2: Process Qualification
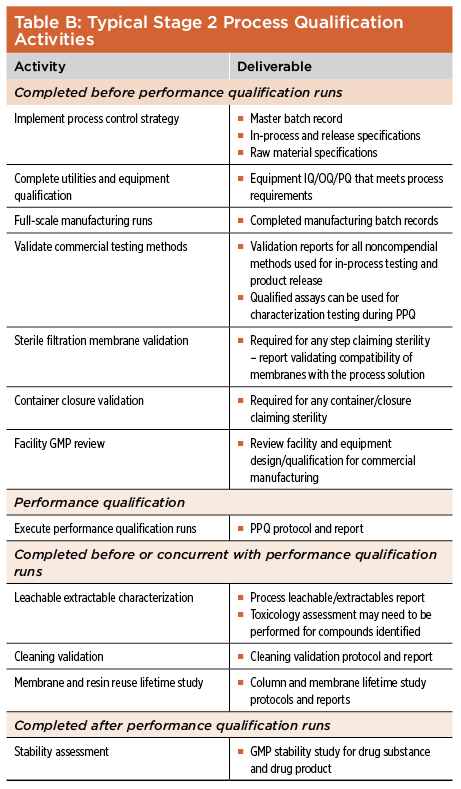
Process qualification, as defined by FDA guidance, shares many of the same features as the traditional fixed-point approach. The main difference is in how the acceptance criteria that define suitability for market registration are set. Process qualification includes an evaluation of the process design defined in Stage 1 to ensure that the manufacturing process is capable of reliably producing a product that meets all release criteria during routine commercial manufacturing. During Stage 2, the defined scaled-up manufacturing process is run at commercial scale by trained staff under full cGMP conditions using prequalified equipment in the proposed commercial manufacturing plant. Complete process qualification will include the validation of the performance of process chemicals and raw materials used in each unit operation, qualification of all supporting facilities and utilities necessary for the manufacturing process, qualification of all process equipment, validation of each individual unit operation, and validation of the entire process as it is intended to be operated at commercial scale. Before process qualification can be performed, a series of related activities outlined in Table B must be completed to ensure the success of the process qualification. These activities include the validation of in-process and release-testing methods, scale-up of the manufacturing process, and validation of related equipment and processes.
Each batch of biopharmaceutical product produced during process qualification is tested using validated in-process and final product test methods to confirm that the product meets preset specifications and in-process acceptance criteria. Additional process characterization methods and analyses are also expected during this stage to fully characterize and qualify the process. Process controls, including the analytical test methods used for both in-process testing and final product release must be sufficient to confirm that each CPP is held within its preapproved range and that the final product meets all release specifications. The combination of process design studies performed during Stage 1 and process qualification performed during Stage 2 should confirm that the various manufacturing processes are reliable, reproducible and that they adequately control all of the product’s CQAs. Assuming this is the case, the process is considered to be “validated” and the product may be released for commercial use.
Stage 3: Process Verification
Following completion of Stages 1 and 2, routine product manufacturing should be monitored using the validated in-process and final product test methods to ensure that the manufacturing process remains in control and that the product continues to meet all CQAs. The particular strategy for continuous process verification in Stage 3 should be dictated by information gathered during Stage 2.21 The intent of this continued process verification is to monitor the process throughout the product life cycle, demonstrating continued control of the manufacturing process. Since changes may occur in the testing protocols or the analytical methods used during the product life cycle, it is important that these revised test methods be appropriately validated and that results of these new methods correlate with those obtained previously. While the FDA guidance does not specify the extent of sampling and testing necessary to ensure adequate process control, it does recommend that monitoring and sampling of process parameters and quality attributes be continued until sufficient data are available to estimate the extent of variability of the manufacturing process. FDA recommends that testing programs be designed by someone with sufficient training and knowledge in statistics to ensure that the monitoring plan meets regulatory expectations and that the overall monitoring plan—including a description of how data trending and all other calculations will be performed—be fully described in the Stage 3 validation protocol.22
The purpose of continued process verification is to establish the appropriate levels and frequency of routine sampling and monitoring for a particular product and process to meet the cGMP requirement of “statistically appropriate and representative levels.”22 During Stage 3, production data should be collected on an ongoing basis and appropriate alert and action limits set. Since the number of batches of biopharmaceutical product produced prior to completion of process qualification (Stage 2) is likely to be small, the amount of sampling and in-process testing required during routine commercial manufacturing may be greater in the early years of commercialization than later in the product life cycle. The data collected should be sufficient to provide strong statistical evidence that all CPPs are being held within their acceptable ranges and that there are no trends among any of the CQAs towards out-of-specification results. As commercial manufacturing progresses, the extent of testing may decrease as increased confidence in process capability and reproducibility is confirmed. Once sufficient data are available to establish the statistically meaningful extent of process variability, the monitoring program can be adjusted accordingly. Continuous process verification strategies will vary from process to process, but typically involves additional process sampling and monitoring outside of parameters routinely recorded in the master batch record. Based on testing results, control ranges for certain operating parameters may be adjusted over time and some routine testing may be eliminated after sufficient manufacturing experience is obtained. The requirements for extensive in-process testing and process monitoring during Stage 3 is more stringent than the simple trending review of routine annual production performance required by regulatory authorities in the past. Once process robustness has been established, some of the extra in-process testing and process monitoring conducted during validation may be discontinued, with appropriate justification.
Defining CPPs
The CQAs of a biopharmaceutical product are those physical, chemical, biological, and microbiological properties and characteristics that must be controlled within an appropriate range to ensure the desired product quality. CQAs are also factors that affect product purity, strength, or stability, particularly post-translational modifications such as glycosylation and heterogeneity resulting from the presence of various glycoforms. The CQAs of a biopharmaceutical product will always include product potency and immunogenicity. Because product-related impurity levels (e.g., aggregated or clipped forms) and other process-related impurities can affect product safety or efficacy, they may also be included in the CQAs for a biopharmaceutical product. A key element of QbD and the new process validation standards is that these CQAs can be linked to certain CPPs in the manufacturing process. These can be identified during the earlier stages of process design by an initial risk analysis, but additional CPPs may be identified at any time during the product life cycle as a result of continuous process monitoring. Besides affecting the CQAs, the ability to control a process parameter within its intended range is a significant factor in defining its criticality, especially in the manufacture of biopharmaceutical products. To control the CPPs for a manufacturing process, it is important to have a clear understanding of the desired settings and ranges for each parameter. During process development, three nested ranges of relevance may be established for each process parameter:
- The widest range is the proven acceptable range (PAR) within which the product produced always meets its desired release specifications and CQAs. Outside the PAR, the process will fail and the product may not meet its desired CQAs. Establishing the PAR is sometimes referred to as “testing to the edge of failure” and is normally done during process development.
- Embedded within the PAR is the regulatory or validated range, which is the range for the parameter that is tested during validation studies and represents those ranges included in a product registration application (e.g., BLA).
- Embedded within the regulatory range is the normal operating range, which is the range for the parameter specified in the master batch record that is expected to be used for routine commercial production of the monoclonal antibody product.
“Pyramiding” ranges for operating parameters (Figure 5) was originally proposed by Chapman in 1984 when he introduced the “proven acceptable range” (PAR) approach to process validation.23 As defined by ISPE, the PAR for a critical parameter is the range determined to be achievable and appropriate for the process or processes with which it is associated.30 –31 By using knowledge gathered during development, the PAR approach helps ensure that the regulatory range for each parameter is wider than the routine operating range and further ensures that the process is not operating at the edge of failure.24 Such an approach allows for minor process variations beyond the operating range, prevents failure of the unit operation or overall process, and results in a more robust process that is less likely to fail.

A risk analysis of each unit operation based on data collected during development and the potential result of failure to control a specific parameter within its acceptable product CQA range should be conducted to establish which of the many process parameters in a biopharmaceutical manufacturing process are critical. This will help refine the acceptable ranges of each parameter and minimize the potential for process variability and failure. Many process parameters in a biopharmaceutical manufacturing process will have wide acceptable ranges, so that it is not necessary to establish what the acceptable range truly is, as long as an operating range is defined within this broad range. These parameters are not likely to be critical. On the other hand, if the PAR for a specific process parameter is narrow, it is likely that parameter is critical to meeting the product CQAs. In such a case, the validated range should be established so that it approaches the boundaries of the acceptable range, but remains safely away from the edge of failure. Both the temperature and pH of the cell culture medium in a bioreactor may have the potential to affect product quality, for example, but the acceptable range for temperature may be relatively broad while the acceptable pH range may be much tighter and represent a much higher risk for product failure resulting from a process excursion outside this range.
Process Validation Planning and Execution
Although it is not mandatory, regulatory agencies have come to expect that a sponsor’s approach to process validation will be described in a validation master plan (VMP). This documents a company’s approach to process validation and also clarifies or defines responsibilities, general objectives, and procedures to be followed for validation. It may reference several protocols, procedures, and processes to qualify different pieces of equipment, and may also specify validation schedules and resource allocations needed to perform each validation study. A typical VMP for the manufacture of biopharmaceutical bulk drug substance should contain, at a minimum, the information listed in Table C.

Individual process validation protocols should:
- Describe the procedures to be followed in detail
- Specify critical and key operational parameters and their respective ranges, as well as data acceptance criteria
- Detail the procedures required to perform the validation, including the sampling plan and the responsibilities of various team members participating in the validation study
- Specify a sufficient number of replicate process runs to demonstrate process reproducibility and provide an accurate measure of variability among successive runs
Test conditions for each process validation run should encompass the upper and lower processing limits and circumstances, including those within standard operating procedures, which pose the greatest chance of process or product failure compared to ideal conditions. Such conditions have become widely known as “worst case” conditions (sometimes referred to as “most appropriate challenge” conditions). The new process validation guidance specifies that it is not necessary to employ the “test-to-failure” approach, but only to ensure that those conditions posing the greatest risk of variation beyond acceptable limits or the greatest risk to the quality of the product should be studied adequately.1 It is anticipated that in the future a design space will be generated for each critical process that encompasses all acceptable operating conditions. At the conclusion of each process validation study, a final validation report should be prepared to documents the results. This report should include data from any qualification or production batch run as part of the protocol, a summary of protocol or batch nonconformances—along with the investigation of the nonconformance and any conclusions or recommendations resulting from the investigations—and a summary of whether the acceptance criteria of the protocol have been met. Aside from meeting the regulatory requirements for process validation, the VMP, validation protocols, and final reports will serve as a repository of key development and process information. These can support future process changes and improvements, as well as further development of the design space for the manufacturing process.
Future Perspective
The increasing adoption and use of manufacturing technology platforms, especially in the production of monoclonal antibodies, and advances in high-throughput automation will continue to strengthen process design and optimization. These advances will expedite the development of high-yielding, reliable, and robust processes.25 ,26 ,27 ,28 ,29 Likewise, continued advances in analytical methods for characterizing biopharmaceutical products and processes, including the development and implementation of process analytical technologies for inline monitoring and control, will provide better and more sophisticated tools to enhance and facilitate process qualification and continuous process verification. In the near term, as industry moves from the traditional fixed-point validation to a life cycle approach, the incorporation of QbD and new concepts of process verification and validation are expected to be flexible as regulatory authorities define the requirements and expectations of these new initiatives. During this transition, regulatory filings are expected to incorporate blended elements of both approaches. In the long run, however, incorporating science-driven risk-based process development and validation will result in more reliable processes that can be readily adapted to new process information. This will ensure continued viability of these processes and minimize the risks of process failures and potential shortages of critical medicines. By conforming to best industrial practices and embracing the new process validation guidelines and initiatives, biopharmaceutical manufacturing will continue to improve for the betterment of our industry and patients worldwide.
By: Francisco C. Castillo, Brendan Cooney, and Howard L. Levine
About the Authors
Frank Castillo, Ph.D., Senior Consultant with BioProcess Technology Consultants, has over 30 years’ experience in the development and manufacturing of biopharmaceuticals with expertise in project management and process development for monoclonal antibodies, recombinant proteins, gene therapy vectors, and cell-based therapies. He was scientific director and head of fermentation and cell culture development at Berlex Biosciences (a subsidiary of Schering AG) and previously held positions at Xoma and the Venezuelan Institute for Scientific Research. He holds a PhD in microbiology from the College of Medicine and Dentistry of New Jersey. Brendan R. Cooney, Associate Consultant of BioProcess Technology Consultants, Inc., has over eight years of experience has a technical and science writer in the pharmaceutical industry and academia. He has been involved in developing and writing document guidance systems and internal documentation systems for both research and commercial applications. Prior to working for BioProcess Technology Consultants, Cooney founded and ran CBRC Consulting, which provided technical writing consulting services to biotech companies including Shire HGT and Histogenics Corporation. Cooney has a B.A. degree from Northeastern University in Boston, Massachusetts where he graduated magna cum laude. Howard L. Levine, Ph.D., President and Principal Consultant of BioProcess Technology Consultants and Managing Director of BPTC Europe, has over 30 years’ experience in the development and commercialization of biopharmaceutical products. He has worked on products including vaccines, cell therapy, fusion proteins, antibodies, natural products, cytokines, and other recombinant proteins. Prior to founding BioProcess Technology Consultants, Dr. Levine was vice president of manufacturing operations at Repligen Corporation and previously held positions of increasing responsibility in process development and manufacturing at Genentech, Amgen, and Xoma. Dr. Levine holds a PhD in chemistry from the University of Chicago and completed a postdoctoral fellowship at Harvard University.
- 25 a b Rameez, S., S.S. Mostafa, C. Miller, and A.A. Shukla. “High-Throughput Miniaturized Bioreactors for Cell Culture Process Development: Reproducibility, Scalability, and Control”, Biotechnology Progress 30, no. 3 (May–June 2014): 718–727.
- 1 a b US Food and Drug Administration. “Guideline on General Principles of Process Validation.” May 1987. www.fda-consultant.com/provalid.html.
- 2———. “Pharmaceutical CGMPs for the 21st Century—A Risk-Based Approach: Final Report.” September 2004. www.fda.gov/downloads/Drugs/DevelopmentApprovalProcess/Manufacturing/QuestionsandAnswersonCurrentGoodManufacturingPracticescGMPforDrugs/UCM176374.pdf
- 3 a b c d International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use. ICH Harmonised Tripartite Guideline Q9: “Quality Risk Management.” Step 4 version. 9 November 2005. www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q9/Step4/Q9_Guideline.pdf.
- 4 a b c US Food and Drug Administration. Guidance for Industry. “Process Validation: General Principles and Practices.” Revision 1. January 2011. www.fda.gov/downloads/Drugs/Guidances/UCM070336.pdf.
- 5European Medicines Agency. “Guideline on Process Validation.” Draft. EMA/CHMP/CVMP/QWP/70278/2012-Rev1. 29 March 2012, www.ema.europa.eu/ema/pages/includes/document/open_document.jsp?webContentId=WC500125399.
- 6Yu, L.X., “Pharmaceutical Quality by Design: Product and Process Development, Understanding, and Control.” Pharmaceutical Research 25, no. 4 (2008): 781–791.
- 7US Code of Federal Regulations. Title 21, Part 820, Section 820.75: “Process Validation.” www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/CFRSearch.cfm?CFRPart=820&showFR=1.
- 8 a b Sidor, L., and P. Lewus. “Validation & Compliance: Using Risk Analysis in Process Validation.” Biopharm International 20, no. 2 (1 February 2007): 25–32.
- 9 a b c d e f CMC Bio Working Group, “A-Mab: A Case Study in Bioprocess Development Version 2.1.” 30 October 2009. Download available at: https://ispe.org/sites/default/files/initiatives/pqli/a-mab-case-study-version.pdf
- 10Burleigh, S.C., et al. “Synergizing Metabolic Flux Analysis and Nucleotide Sugar Metabolism to Understand the Control of Glycosylation of Recombinant Protein in CHO Cells.” BMC Biotechnology 11, no. 95 (18 October 2011). bmcbiotechnol.biomedcentral.com/articles/10.1186/1472-6750-11-95.
- 11Kunkel, J.P., et al. “Comparisons of the Glycosylation of a Monoclonal Antibody Produced Under Nominally Identical Cell Culture Conditions in Two Different Bioreactors.” Biotechnology Progress 16, no. 3 (May-June 2000): 462–470.
- 12Katz, P., and C. Campbell. “FDA 2011 Process Validation Guidance: Process Validation Revisited.” Journal of GXP Compliance 16, no.4 (Autumn 2012): 18–29.
- 13International Electrotechnical Commission. International Standard IEC 60812. “Analysis Techniques for System Reliability – Procedure for Failure Mode and Effects Analysis (FMEA).” 2nd edition, 2006.
- 14US Food and Drug Administration. “HACCP and Application Guidelines.” 14 August 1997. www.fda.gov/Food/GuidanceRegulation/HACCP/ucm2006801.htm.
- 15Boychyn, M., R. Hart., and K. Frandsen. “Design of a Viral Contamination Barrier for a Serum-Containing Cell Culture Process.” Presented at Bioprocess International Conference, Providence, Rhode Island, 22 September 2010.
- 16Nosal, R., T. Shultz. “PQLI Definition of Criticality,” Journal of Pharmaceutical Innovation 3, no. 2 (June 2008): 69–78.
- 17International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use. ICH Harmonised Tripartite Guideline Q8(R2): “Pharmaceutical Development.”2005. www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q8_R1/Step4/Q8_R2_Guideline.pdf.
- 18Parenteral Drug Association. “Process Validation: A Lifecycle Approach.” Technical Report 60. 2013.
- 19 a b European Medicines Agency, and US Food and Drug Administration, “EMA-FDA Pilot Program for Parallel Assessment of Quality-by-Design Applications: Lessons Learnt and Q&A Resulting from the First Parallel Assessment.” 20 August 2013. www.ema.europa.eu/docs/en_GB/document_library/Other/2013/08/WC500148215.pdf.
- 20Joneckis, C. “Regulatory Expectations for Process Validation.” Presented at the California Separation Science Society’s Well Characterized Biological Products (WCBP) Conference. Held 6–10 January 2014.
- 21McNally, G.E. “Process Validation: A Lifecycle Approach.” 6 May 2011. www.fda.gov/downloads/AboutFDA/CentersOffices/CDER/UCM255585.pdf.
- 22 a b Chapman, K.G., “The PAR Approach to Process Validation,” Pharmaceutical Technology 8, no. 12 (December 1984): 22–36.
- 23Chapman, K.G., et al. “Proposed Validation Standard VS-1.” Journal of Validation Technology 6, no. 2 (February 2000): 502–521. www.readbag.com/gxpandjvt-ivtnews-pdf-pharmstandards
- 30International Society for Pharmaceutical Engineering. Baseline Guide Volume 2: Oral Solid Dosage. 2nd edition. November 2009. www.ispe.org.
- 31———. “Proven Acceptable Range.” ISPE Glossary of Pharmaceutical and Biotechnology. www.ispe.org/glossary?term=Proven+Acceptable+Range+(PAR).
- 24Greb. E., and A. Drakulich. “Platform Technologies.” Pharmaceutical Technology 36, no. 3 (March 2012): 46–52
- 26Bhambure, R., K. Kumar, and A.S. Rathore. “High-Throughput Process Development for Biopharmaceutical Drug Substances. “Trends in Biotechnology 29, no. 3 (March 2011): 127–135.
- 27Chollangi, S., et al. “Accelerating Purification Process Development of an Early Phase MAb with High-Throughput Automation: Part 1,” Bioprocess International 12, no. 3 (March 2014): 48–52. www.bioprocessintl.com/wp-content/uploads/bpi-content/BPI_A_141203AR06_O_231757a.pdf.
- 28Chollangi, S., et al. “Accelerating Purification Process Development of an Early Phase Mab with High-Throughput Automation.” Bioprocess International 12, no. 4 (April 2014): 32–41. www.bioprocessintl.com/2014/accelerating-purification-process-development-of-an-early-phase-mab-with-high-throughput-automation-351110
- 29International Society for Pharmaceutical Engineering. Baseline Guide Volume 2: Oral Solid Dosage. 2nd edition. November 2009. www.ispe.org.