Accommodating Multiple Modalities in the Same Facility

Many organizations are evaluating how advanced therapy medicinal products (ATMPs) and other traditional modalities may be combined within the same facility or within a newly constructed agnostic building. This article outlines a broad framework to evaluate different types of modalities that may be accommodated concurrently in a new or existing facility and then uses a case study to explain how the approach may be applied to an existing facility.
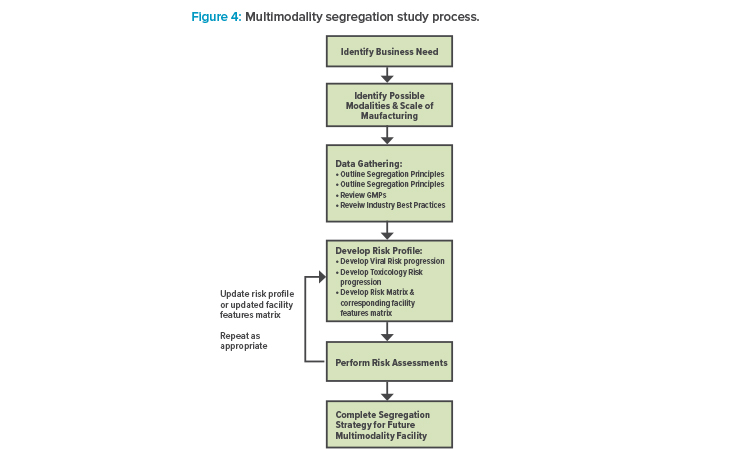
ATMP facilities are different from conventional pharmaceutical facilities that process other traditional modalities, such as vaccines and monoclonal antibodies (mAbs), and often require heightened segregation and smaller footprints. A new framework is proposed to accommodate multiple different modalities, with six major steps: (1) identify the business need; (2) identify possible modalities and the scale of manufacturing; (3) complete data gathering, which includes outlining segregation principles and reviewing GMPs and industry best practices; (4) develop a risk profile, which includes developing viral and toxicity risk progression, a risk matrix, and a facility features matrix; (5) perform risk assessments, and then update the risk profile and facility features matrix as needed; and (6) complete a segregation strategy for a future multimodality facility.
Business Need, Possible Modalities, and Manufacturing Scale
When a business need is being developed, it must be determined which modalities are of interest to the organization, how their manufacturing might be accommodated, and at what scale they should be manufactured. These modalities may include cell therapies, gene therapies, tissue engineering, nucleic acid synthesis, vaccines, and mAbs.
Complete Data Gathering
Outline Guiding and Segregation Principles
Next, broad guiding principles and key segregation principles should be determined as part of the data-gathering step. Key segregation principles are required for existing, proposed, and potential new modalities. Some example principles and assumptions that should be determined and agreed upon relate to facility design, manufacturing area capacity, defining process steps, safety requirements, and infrastructure.
Facility design: All new facilities will be designed and built to current industry best practices using the latest available technology. More specifically, closed systems, isolators, and single-use technology are to be adopted as appropriate. Alternatively, an existing facility will be agile and can facilitate early-stage processes received through acquisition. These processes may still have open aseptic steps. The manufacturing spaces should be able to accommodate the full journey through to process closure.
Manufacturing area capacity: The capacity of the individual manufacturing areas should be established. This may include answering questions like: Is the facility aimed at commercial manufacturing? Is the facility designed only for volumes to support clinical trials?
Defining process steps: The core processing steps for each modality should be defined. If processes are not already fully defined, the assessment will need to make broad assumptions in terms of processing steps and possible technologies to be used. However, future assessments will be required to either confirm or modify some of the assumptions based on actual product and process information. Any decisions or recommendations are ultimately determined by quality risk management (QRM) principles and practices, as well as relevant GMPs.
Safety requirements: Similar to GMP requirements (see next section), all safety requirements should be agreed upon. This should include biosafety, hazardous agents, and any occupational hygiene considerations. This will require broad assumptions and understanding of some of the modalities being considered. For the purposes of brevity, this article doesn’t discuss safety. However, it is noteworthy that often safety and GMP requirements drive complementary results.
Infrastructure: All available infrastructure that will be used should be considered; for example, shared quality control laboratories, utilities, and waste handling.
Review Guidance
Regulations
The assessment must outline which GMPs are being used. For example, an assessment for a facility primarily focused on US and EU markets would only consider GMPs and regulations from EudraLex and the FDA. However, with an agnostic building, the breadth of regulations that must be considered can be extensive.
The following list includes regulations that can be considered, but it is by no means exhaustive: EudraLex Volume 4 Parts I, II, and IV and Annexes 1 and 2;2 , 3 , 4 , 5 ,6 EU human tissues and cells directive 2004/23/EC;7 FDA CFR Title 21 Parts 211, 600, and 1271;8 ,9 , 10 FDA Guidance for Industry: Sterile Drug Products Produced by Aseptic Processing–Current Good Manufacturing Practice,11 and Guidance for Industry: Current Good Tissue Practice (CGTP) and Additional Requirements for Manufacturers of Human Cells, Tissues and Cellular and Tissue-Based Products (HCT/Ps).12 Also, regulatory agencies may refer to a corresponding pharmacopeia such as USP 800.1
These regulations contain some requirements that relate to specific modalities. For penicillins and cephalosporin (highly sensitizing materials) and for pathogenic organisms (i.e., Risk Group 3 or 4), dedicated and self-contained facilities that include air-handling equipment and process equipment should be employed.3 In addition for penicillins specifically, air-handling systems for manufacture, processing, and packing shall be completely separate from those for other drug products for human use.8
It is possible to develop a basic set of guidelines to accommodate multiple modalities within the same facility once that facility’s main purpose has been established. But to do so, it is important to understand what boundaries may exist.
For materials of an infectious nature (Risk Group 1 and 2) or with high pharmacological activity or toxicity (such as certain steroids or cytotoxic anti-cancer agents), dedicated facilities are not required. However, these do require that validated inactivation and cleaning procedures able to reduce product residues to amounts below defined safety thresholds are established and maintained.3 For live vaccines, manufacturing in a separate wing of a building is acceptable, with the appropriate controls to prevent cross-contamination of other products within the building.9
If the manufacturing site produces medicinal products other than ATMPs, based on a risk assessment, ATMP manufacture may need to take place in a dedicated area of the facility. Special precautions should be taken in the case of manufacturing activities involving infectious viral vectors (e.g., oncolytic viruses): these activities should take place in a segregated area.4
Industry Best Practices
In addition to regulations, there are many industry best practices that further interpret GMPs. Some practices are recorded in industry best practice guides and can include ISPE Guides, Baseline Guides, and Good Practice Guides, and Parenteral Drug Association (PDA) technical reports.13 ,14 ,15 Other industry best practices may not be formally documented in regulations or industry best practice guides, but should also be investigated as part of the assessment.
The assessment may also identify perception concerns. Owner companies—they own both the product and the manufacturing facility—will have freedom to set their own segregation principles. For example, a contract manufacturing organization (CMO) that may have differing client modalities adjacent to each other will want a segregation policy that is acceptable to a wider customer base.
| Low Risk | Medium Risk | High Risk | Very High Risk |
|---|---|---|---|
Caustic, acids, medias, salts, and other commodity chemicals (< OEB 2) For example:
|
Manufacture of cytotoxic products (≤ OEB 3)2 For example:
|
Manufacture of cytotoxic products (> OEB 3)1,2 For example:
|
Sensitizing agents with specific requirements mentioned in regulations For example:
|
1 Carcinogenic, mutagenic, and teratogenic materials need to be considered on a case-bycase basis. In general, any use of these materials will be in small volumes and will receive dedicated risk assessments.
2 OEB classification to include assessment of carcinogenic, mutagenic, and reproductive (CMRs) toxic substances.
Internal Corporate Standards
The manufacturer may have internal corporate standards that address how any manufacturing asset should be designed and operated irrespective of location or markets served. These standards should be listed prior to commencing this exercise.
Develop a Risk Profile
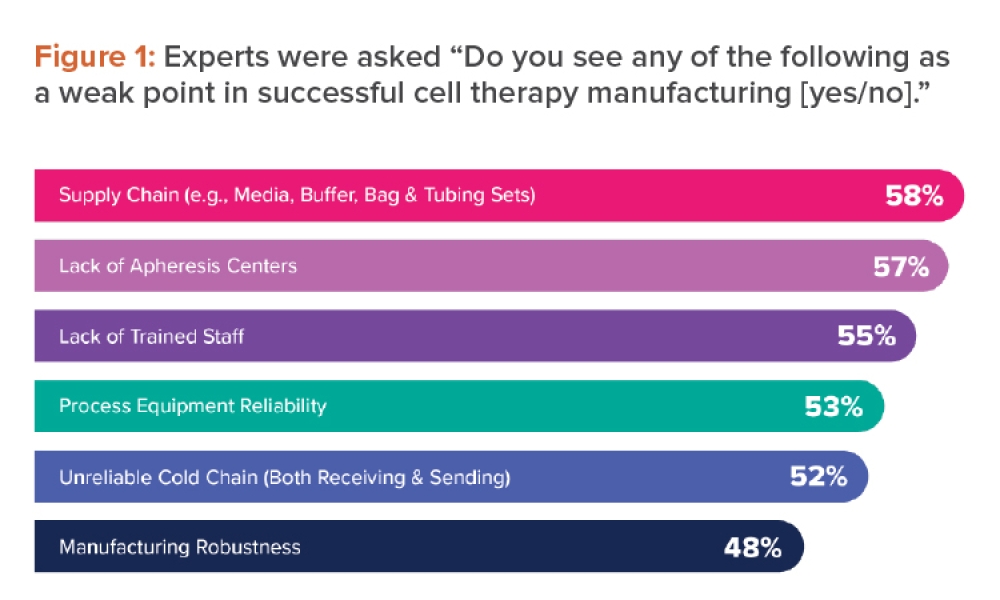
It is necessary to develop a risk profile for different modalities to establish some key segregation principles. The risk profile should also be based on regulations, industry best practices, and the company’s own risk profile and practices. In this article, two separate risk profiles have been developed: chemical potency and toxicity risk, and biological (viral and microbial) risk.
In both cases, the category of risk ranges from low to very high. Each risk profile category contains a definition with examples. Profiles also form the basis for the proposed segregation principles in the case study. Very-high-risk profiles align with clear regulatory expectations for dedicated manufacturing facilities.
Chemical Potency and Toxicity Risk Progression
In Table 1, the authors have captured some types of biotechnology modalities that relate more specifically to potency and toxicity with respect to risk profile: low, medium, high, and very high risk. The potency ranges from occupational exposure band (OEB) 1 to 5, with 5 being the highest potency. The final risk profile is determined by the toxicity and potency data, containment strategy, and technology used.
Cleaning of shared equipment for multiproduct facilities is a major concern in GMPs. In addition, the HVAC, utilities, material, waste, and personnel flows all must be assessed. The toxicity assessment feeds into all of these considerations. The QRM approach focuses on how cross-contamination can occur across modules through HVAC, utilities, material, waste, and personnel flows. For example, if an isolator is capable of containing an OEB 5 process, the operator may only be exposed to microgram levels. Therefore, a risk assessment will look at how many micrograms of material can contaminate a different process. More specifically, it will ascertain if the level of theoretical cross-contamination exceeds the permitted daily exposures (PDE) for a product in another module.
Risk-Based Manufacture of Pharmaceutical Products
A useful guide for performing the risk assessments is the ISPE Risk-Based Manufacture of Pharmaceutical Products Baseline Guide, often referred to as Risk-MaPP. 13 The second edition of the ISPE guide received regulatory input from the FDA and other agencies and was published in 2017. The guide has a useful workflow for assessing multiproduct manufacturing and advocates the QRM approach. The four key areas in the Risk-MaPP approach, in the order of decreasing risk, are as follows: mix up, retention (carry-over), mechanical transfer, and airborne.
Mix up: Mix up refers to the general tracking and control of materials. For example, materials may be closed on delivery to dedicated suites, i.e., not opened adjacent to other materials. Robust wipe-down procedures and industrial hygiene practices apply at all times. Control storage may be necessary in the warehouse, along with segregated waste flow as required.
Retention (carryover): Modules and associated equipment may be dedicated to a product with no carryover risk from equipment cleaning. If using single-use technology, then there is minimum risk from cleaning. In-suite cleaning facilities may be required. Inactivation methods for active pharmaceutical ingredient (API) and organisms or DNA and RNA may also be required in the suites. All potential leaks, spills, and material air lock (MAL) operations will need to be risk assessed.
Mechanical transfer: Mechanical transfer often refers to operators and how they may become contaminated with product and then inadvertently contaminate another person, product, or both. The use of isolators and/or closed processes—along with a suitable gowning policy and layout (e.g., unidirectional flow of personnel, product, and waste, as required)—helps reduce the risk of contamination. Dedicated procedures for spills or deactivating an operator gown when soiled may be required.
Airborne: A “sink” or “bubble” concept for airlocks will limit the airborne transfer to corridors. There may also be a requirement for a segregated HVAC unit with or without terminal HEPA filtration on the supply and return.
The Risk-MaPP approach is a useful guide that will help risk assess products and processes based on QRM principles.
High-Potency and High-Toxicity Products to Be Excluded
From a regulatory perspective, the only products that are to be excluded are sensitizing agents, including penicillins and cephalosporins (beta-lactams); potentially genotoxic compounds; and potentially OEB 5 compounds where the risk assessment indicates the compound cannot be adequately controlled. Manufacturing of (or with) high-potent compounds in adjacent suites may be possible with the appropriate engineering controls and risk assessments based on toxicity data.
Biological (Viral and Microbial) Risk Progression
There is not a single document for assessing different modalities where varying levels of viral and microbial risks are present equivalent to the ISPE Risk-MaPP guide for chemical potency and toxicity risk. The PDA publications “Virus Contamination in Biomanufacturing: Risk Mitigation, Preparedness, and Response” 14 and “Points to Consider for Microbial Control in ATMP Manufacturing” 15 are very useful publications that should be read as part of this exercise.
These documents outline practical engineering and procedural controls, which need to be used for mix modality manufacturing; however, these documents are written from the perspective of an adventitious viral or microbial contamination from external environments and raw materials. Similarly, closed processing risk assessment methodologies are available (such as the ISPE Biopharmaceuticals Manufacturing Facilities Baseline Guide), but these are written in the context of multiproducts within mAb or therapeutic protein where viral risk is uniform across the processes considered.
Therefore, a science- and risk-based approach from first principles must be taken to assess biological risk. QRM tools commonly used in the biopharma industry should provide good guidance on what is acceptable. Table 2 captures the types of modalities that relate more specifically to viral and microbial organisms with respect to risk profile, and provides an approximate risk ranking of viral and microbial risks. It can be used as an initial guide until the actual level of risk is determined by a more comprehensive risk assessment. A site biosafety committee will be required to help categorize the risk grouping of hazardous organisms.
Understanding the Biological Risk Categories
Viral and microbial products have been classified into the following broad categories (excluding very high risks that are not in scope): low, medium, and high risk (very high risk is not discussed here). Low risk includes mAb processes where characterized murine cell banks are used, BSL-1 bioprocesses, and fill finish of traditional therapeutic proteins. These are low-risk processes because the presence of viral material is low and if it is present, it is likely to be of animal origin and are not known to propagate in humans. Medium-risk processes include those that use viral vectors, human cell lines where an adventitious human virus will replicate, or animal tissue that may have viruses but those that are not known to propagate in humans. High-risk processes are typically those where human viruses may be present. These include human-donated material and culturing of live human viruses.
| Low Risk | Medium Risk | High Risk | Very High Risk |
|---|---|---|---|
This includes established and controlled practices, including the use of controlled animal-derived media components |
|
These processes have specific guidelines within regulations |
|
Based on this risk categorization, the low-risk and high-risk processes are easier to distinguish. Medium-risk processing covers a broad middle ground and careful consideration of the underlying science will be needed in the corresponding risk assessments. It may result in the process being reclassified as either low or high risk. In general, it is possible to conclude that medium-risk viral and microbial processes may be accommodated within the same facility, provided there is data to support a comprehensive risk assessment.
The following have been categorized as medium-risk processes: human cell lines, allogeneic cell therapy (screened cell stock), processing of proteins extracted from animal sources (e.g., heparin, insulin); replication incompetent viral vector/gene therapy; and viral antigen vaccines. The overarching principle is that although human cells are often being cultured, their origin is known or, better still, characterized. Therefore, in principle, there are no viruses present, but care is taken for adventitious viruses. The level of screening of donors will be key to the risk characterization of this type of processing. If inadequate screening is performed, this may be recategorized as high risk. Similarly, viral vectors, gene therapies, and viral antigen vaccines have viral properties. However, they have been genetically engineered to be replication incompetent outside of the bioreactor environment. Many of these examples fall into the category of ATMPs. Similar to the high-potent compounds, there may be different and conflicting perceptions that will need to be managed. It may be more prudent to consider fully segregating these types of processes.
- 2European Commission. “EudraLex, Volume 4: EU Guidance for Good Manufacturing Practices for Medicinal Products for Human and Veterinary Use. Part I: Basic Requirements for Medicinal Products.” Published 2003. https://health.ec.europa.eu/medicinal-products/eudralex/eudralex-volume-4_en#part-i---basic-requirements-for-medicinal-products
- 3 a b c European Commission. “EudraLex, Volume 4: EU Guidance for Good Manufacturing Practices for Medicinal Products for Human and Veterinary Use. Part II: Basic Requirements for Active Substances Used as Starting Materials.” Published 2014. https://ec.europa.eu/health/sites/health/files/files/eudralex/vol-4/2014-08_gmp_part1.pdf
- 4 a b European Commission. “EudraLex, Volume 4: EU Guidance for Good Manufacturing Practices for Medicinal Products for Human and Veterinary Use. Part IV: GMP Requirements for Advanced Therapy Medicinal Products.” Published 2017. https://health.ec.europa.eu/system/files/2017-11/2017_11_22_guidelines_gmp_for_atmps_0.pdf
- 5European Commission. “EudraLex, Volume 4: EU Guidelines for GMPs for Medicinal Products for Human and Veterinary Use. Annex 1: Manufacture of Sterile Medicinal Products.” Published 2008. https://ec.europa.eu/health/sites/default/files/files/gmp/2017_12_pc_annex1_consultation_document.pdf
- 6European Commission. “EudraLex, Volume 4: EU Guidance for Good Manufacturing Practices for Medicinal Products for Human and Veterinary Use. Annex 2: Manufacture of Biological active substances and Medicinal Products for Human Use.” Published 2007. https://health.ec.europa.eu/system/files/2019-02/2018_annex2_en_0.pdf
- 7European Commission. “Directive 2004/23/EC of the European Parliament and of the Council of 31 March 2004 on Setting Standards of Quality and Safety for the Donation, Procurement, Testing, Processing, Preservation, Storage and Distribution of Human Tissues and Cells.” Published March 2004. EUR-Lex - 32004L0023 - EN - EUR-Lex (europa.eu)
- 8 a b US Food and Drug Administration. “CFR Title 21 Part 1271: Human Cells, Tissues, and Cellular and Tissue-Based Products.” Published January 2001. https://www.ecfr.gov/current/title-21/chapter-I/subchapter-L/part-1271
- 9 a b US Food and Drug Administration. “CFR Title 21 Part 211: Current Good Manufacturing Practice for Finished Pharmaceuticals.” Published September 1978. https://www.ecfr.gov/current/title-21/chapter-I/subchapter-C/part-211
- 10US Food and Drug Administration. “CFR Title 21 Part 600: Biological Products: General.” Published March 2005. https://www.ecfr.gov/current/title-21/chapter-I/subchapter-F/part-600
- 11US Food and Drug Administration. “Guidance for Industry: Sterile Drug Products Produced by Aseptic Processing–Current Good Manufacturing Practice.” Published October 2004. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/sterile-drug-products-produced-aseptic-processing-current-good-manufacturing-practice
- 12US Food and Drug Administration. “Current Good Tissue Practice (CGTP) and Additional Requirements for Manufacturers of Human Cells, Tissues and Cellular and Tissue-Based Products (HCT/Ps).” Published December 2011. https://www.fda.gov/media/82724/download
- 1US Pharmacopeial Convention. “USP<800>: Hazardous Drugs—Handling in Healthcare Settings.” Rockville, MD: US Pharmacopeial Convention, 2016.
- 13 a b International Society for Pharmaceutical Engineering. ISPE Baseline Guide, Vol. 7, Risk-Based Manufacture of Pharmaceutical Products, 2nd ed. North Bethesda, MD: International Society for Pharmaceutical Engineering, 2017.
- 14 a b Parenteral Drug Association. “Virus Contamination in Biomanufacturing: Risk Mitigation, Preparedness, and Response.” Technical Report No. 83. Published 2019. https://www.pda.org/bookstore/product-detail/5139-tr-83-virus-contamination-in-biomanufacturing
- 15 a b Parenteral Drug Association. “Points to Consider for Microbial Control in ATMP Manufacturing.” Published 2022. https://www.pda.org/bookstore/product-detail/6754-ptc-for-microbial-control-in-atmp-manufacturing
High-risk viral and microbial processes will invariably face regulatory or perception concerns. For example, CAR-T manufacturing acknowledges that patient cells arriving into the facility may harbor infectious biohazardous organisms. As a result, dedicated material flows for patient material arriving and leaving the facility is common. This will be difficult to achieve in many facilities unless it is initially included in early design stages. Similarly, live vaccine manufacturing and any blood processing would require the acknowledgment of a high possibility or intentional presence of human viruses.
Biological Products to Be Excluded
From a regulatory perspective, the only products that should be excluded are spore-forming microorganisms, due to their persistence in the cleanroom environment. Processing products at BSL-3 and BSL-4 should be excluded and there is strong consideration against manufacturing of live vaccines.
Develop a Risk Matrix
Once the risks for each category have been determined, a risk matrix should be completed to align the team with the escalating risks from differing modalities (see Figure 1). Although this is an important step in the mix modality risk assessment and the risk matrix is a useful guide, the assessment team must evaluate it themselves to calibrate their own risk appetite and ensure that particular modalities of interest are properly characterized. It cannot be sufficiently stressed that the underlying science of a particular modality should be understood when characterizing the matrix info. New modalities or variations of existing modalities are constantly appearing, so evaluating the matrix for each new project is important.
Develop a Facility Features Matrix
An agnostic facility is a pre-invested facility that has been designed to accommodate a range of manufacturing processes. It is typically constructed with the core internal process area left unconstructed. This allows a rapid buildout of the manufacturing area once a business need is identified and greatly reduces deployment times.
Because of this, an agnostic facility may have multiple manufacturing spaces, each capable of being fitted out for working with a particular modality. These spaces may be spread across a specific manufacturing level or across multiple floors.
When assessing the adjacency of different modalities, key considerations will include the equipment and closure design (primary containment), whether a single or separate supply and return corridor is required, and whether physical or temporal product segregation is needed. Secondary containment factors such as the HVAC design, pressurization regime, and airlock design will also need to be considered. The segregation of air between manufacturing spaces will be input into any analysis.
It should be noted that if one of the spaces is assigned a high-risk modality and the others are low risk, many of the engineering and architectural controls to minimize cross-contamination will be driven by the highest risk modality.
As a generalization, the following will apply when choosing to add a higher-risk modality: increased secondary containment (e.g., airlocks and pres-sure cascades); progressively stricter personnel, material, and waste flows with consideration for supply and return corridors; potential for dedicated locker rooms; increased reliance on closed processing, barrier technology for open steps, and physical segregation compared to temporal segregation; and less reliance on procedures and training.
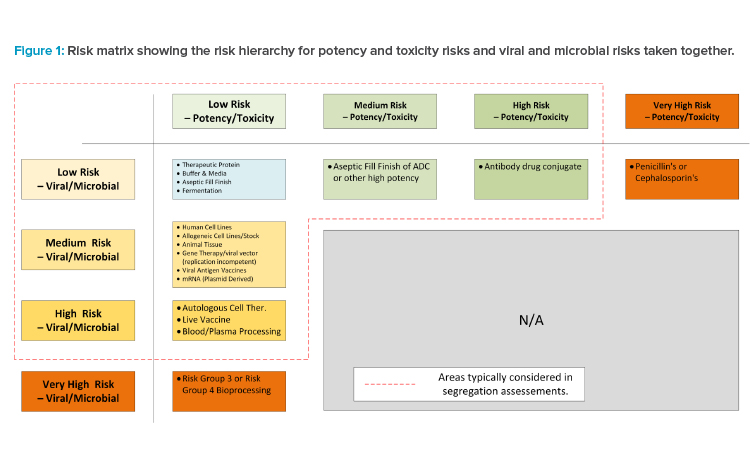
Determine Requirements by Risk Group
Once the core risk groups have been developed, the assessment team must now assess the corresponding facility features for each risk group. This exercise should propose the various facility features. Figure 2 outlines a high-level example, giving some cases of escalating design features and possible in-flection points where facility systems can no longer be shared.
Figure 2 is simply the starting point for a GMP risk assessment for the proposed modalities the manufacturer intends to operate adjacent to each other. Once this matrix has been developed, GMP risk assessments should be performed with a cross-functional team to ensure that the level of segregation is sufficiently robust.
Perform Risk Assessment and Complete Segregation Strategy
Risk assessments should be performed for potential failures that may occur in various manufacturing areas and any connected pieces of infrastructure (e.g., corridors, utilities, and HVAC). These risk assessments should be supported with typical process descriptions and a very high-level layout based on the features described previously. The study should perform risk assessment on possible failures that may lead to cross-contaminations in the manufacturing spaces. The following are examples of typical failures:
Failure 1: Operator wearing a soiled gown, after a spill, is returning to the locker room and passes by personnel entering other manufacturing spaces.
Failure 2: Waste containers are not sealed correctly and leak waste material onto the floor of the corridor.
Failure 3: HVAC failure in manufacturing space #3 causes risk of air mixing between manufacturing spaces, which leads to a cross-contamination.
There are many failure modes that can be considered and these examples represent only a selection. The risk assessment may conclude that some of the escalating features outlined in need to be adjusted to reduce the risk associated with various failure modes.
Once the risk assessment is complete, the risk profile and facility features matrix should be updated as needed, with this process being repeated as appropriate. Finally, a segregation strategy for a future multimodality facility should be completed.
Biological Manufacturing CMO Case Study
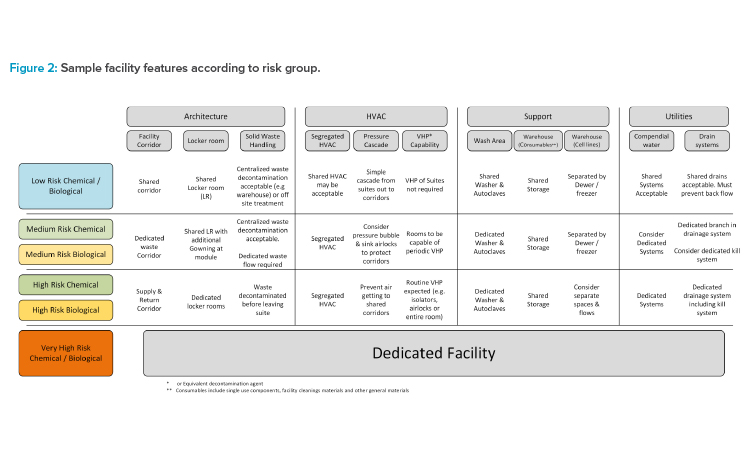
This case study references a CMO whose primary business is focused on biological products. This CMO has constructed a large agnostic facility with pre-invested infrastructure, e.g., clean utilities. The facility consists of multiple manufacturing floors with a just-in-time (JIT) warehouse and support areas, including waste management on the ground floor. Material is supplied to each floor via an elevator from the ground floor JIT warehouse, with further elevators included for separate waste streams. Each floor is designed to accommodate multiple modalities’ manufacturing processes (see Figure 3). The manufacturer concluded that modalities of the same risk group will be shared within manufacturing floors. This will allow common approaches to personnel flows, gowning, and airflows.
For low-risk modalities, these can share a loop corridor because waste material can be easily contained prior to leaving the manufacturing space. It was determined that all manufacturing spaces should have unidirectional flow within the space, but the corridor can be one corridor. It must be noted that the loop corridor does permit the majority of waste and material flows not to cross due to the position on the corridor of the materials lift and waste lifts. Once personnel have entered a manufacturing space, they will not be permitted to enter other space without returning to the locker room (which is access controlled using swipe cards). The manufacturer decided that each room has its own dedicating air handlers with terminal HEPA filters and the common corridor has its own air handler. Because the risk is low, the pressure cascade is simply from the suites to the common corridor.
The medium-risk modalities considered (viral vector, viral antigen vaccines, and allogeneic cell therapy) will require a waste corridor because the risk of cross-contaminating with viral material was considered too high. Similarly, badge access control would limit personnel movement between areas. Each room has its own air handler dedicated to each suite. However, the pressure cascades: the supply-side airlocks operate as pressure sinks to prevent air getting to the common supply corridor where operators from different suites can mix.
High-risk manufacturing modalities are isolated and multiple higher-risk modalities do not share locker rooms. It was determined that one manufacturing process should use the entire floor and not have multiple higher-modality processes adjacent to each other. If the floor was too large for a desired modality (e.g., autologous CAR-T), then the process should be accommodated elsewhere in a dedicated building. As there is only one suite on a floor, the HVAC strategy can be simpler, with a dedicated air-handling strategy for this floor.
Naturally, the preceding overview is quite simplified for the purposes of this article, with many details excluded for brevity. The key point is noting which higher-risk scenario engineering controls must increase to mitigate higher risk. A completed assessment should conclude with architectural layouts, including area classifications, personnel, materials flows, and HVAC zone and pressurization drawings. The assessment must also include warehouse and quality control areas that will share areas and functions.
Conclusion
It is possible to develop a basic set of guidelines to accommodate multiple modalities within the same facility once that facility’s main purpose has been established. But to do so, it is important to understand what boundaries may exist. For example, there is clear regulatory guidance on which modalities require separate facilities. The approach outlined in this article identifies some basic guiding principles, working within the regulations that apply, and develops a basic risk profile to determine how best to accommodate the different modalities given any facility constraints that may exist (see Figure 4).
The proposed six-step approach is designed to help understand which modalities may be suitable for a given facility, and how those modalities may be accommodated. Although each facility may be different, with a different set of recommendations, the described approach can be used during early feasibility. It is worth noting that, as with all early feasibility studies, it is important to reassess the outcome and recommendations as more concrete process information becomes available and as QRM is applied.